
Abstract
Tuberculosis (TB) is a contagious disease that predominantly affects the lungs, but can also spread to other organs via the bloodstream. TB affects about one-fourth population of the world. With age, the effectiveness of Bacillus Calmette-Guérin (BCG), the only authorized TB vaccine, decreases. In the quest for a prophylactic and immunotherapeutic vaccine, in this study, a hypothetical mRNA vaccine is delineated, named MT. P495, implementing in silico and immunoinformatics approaches to evaluate key aspects and immunogenic epitopes across the PstS1, a highly conserved periplasmic protein of Mycobacterium tuberculosis (Mtb). PstS1 elicited the potential to generate 99.9% population coverage worldwide. The presence of T- and B-cell epitopes across the PstS1 protein were validated using several computational prediction tools. Molecular docking and dynamics simulation confirmed stable epitope-allele interaction. Immune cell response to the antigen clearance rate was verified by the in silico analysis of immune simulation. Codon optimization confirmed the efficient translation of the mRNA in the host cell. With Toll-like receptors, the vaccine exhibited stable and strong interactions. Findings suggest that the MT. P495 vaccine probably will elicit specific immune responses against Mtb. This mRNA vaccine model is a ready source for further wet-lab validation to confirm the efficacy of this proposed vaccine candidate.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) is a transmissible disease that is characterized by the infection of the opportunistic bacterial species Mycobacterium tuberculosis (Mtb), primarily targeting the lung [1, 2]. TB is initiated when Mtb is deposited onto the surface of the lung alveoli from the airborne droplets containing the pathogen. Pathogen-containing droplets are mostly dispersed by people with active pulmonary or laryngeal TB. Inhalation of these droplets results in symptoms such as persistent cough, fever and night sweats [3, 4].
The most common cause of mortality caused by a single infectious agent is TB [5]. In 2020, 1.44 million individuals died, with 214,000 of them being HIV-positive [6]. Around 1.7 billion people were latently infected with Mtb in 2014, almost a quarter of the world population [7]. Generally, individuals with a healthy immune system can suppress the growth of Mtb, but the situation gets complicated for immunocompromised patients (e.g. HIV infected or patients with diabetes) who cannot generate enough immune response to suppress the progression of infection [1, 4, 5, 8].
Pulmonary macrophages play a critical role in the primary immune response against Mtb upon entry while a minimum of 12 days are required for the CD4+ T-cells after aerosol infection to respond [9,10,11]. During this period, Mtb increases its population number by > 20,000-fold [9]. Mtb is readily phagocytized by the macrophages present in alveoli. Most often the entering bacteria are killed by these macrophages. But some bacteria can escape from being phagocytized and they start to proliferate within the macrophage as an intracellular parasites [10].
To provide better protection against Mtb, vaccination is a must as the adaptive immune system requires a lot of time to be activated against this pathogen. Moreover, studies on Mtb infected patients have revealed the presence of various multidrug-resistant strains as well as extensively drug-resistant strains [12]. Currently, the only available approved vaccine against TB is Bacillus Calmette-Guérin, BCG [12, 13]. BCG is generally injected into newborn babies which protects them from Mtb till the age of 10 years [8]. For adults, the efficacy of the BCG vaccine varies greatly between 0 and 80% [12]. Several vaccines are in clinical trial, showing promising results. For instance, the M72/AS01E vaccine, a subunit vaccine, is showing very good results in its clinical trial [14]. Although there are several vaccines in the clinical trial phase, currently there is no mRNA-based vaccine developed for TB, neither in the clinical trial phase nor licensed.
mRNA vaccines are a rapidly develo** area. A vast amount of preclinical evidence has been obtained recently, and several human clinical trials have begun [15]. Based on this evidence, mRNA is now considered a safe and effective alternative to (subunit) protein, chimeric virus, and even DNA-based therapies in the form of vaccination [16]. The transient expression and accumulation of selected antigens in the cytoplasm are induced by mRNA.
The proteasome in the cytoplasm of antigen-presenting cells (APCs) can breakdown the antigen into peptides. In the endoplasmic reticulum, the peptides with antigenic properties, are complexed with nascent major histocompatibility complex (MHC) class I molecules. The peptide–MHC complexes can then activate CD8+ cytotoxic T (Tc) cells when they are expressed on the surface of the cell membrane of APCs. CD4+ helper T (Th) cells are activated by MHC class II–peptide complexes expressed on the surface of the cell membrane of APCs. Antigens, secreted by, or released from dead cells that have uptaken and translated the exogenous mRNA, can bind with B-cells within the extracellular matrix, activating these cells. As a result, all adaptive immune effectors, including B lymphocytes, Tc-cells and Th-cells, will be activated by mRNA-based vaccines [16, 17].
In silico analysis of target proteins has simplified the identification of immunogenic B- and T-cell epitopes in the proteins, facilitating the detection of the antigenic epitopes specifically having the potential of eliciting an immune response in particular [18]. Because in silico predictions can minimize the number of experiments required, this strategy is both cost-effective and convenient [19, 20]. However, in silico vaccine design strategy seems to be quite effective, but it might not be efficient enough to catch pace with the advent of newer pathogens. All findings must be thoroughly and extensively analyzed in order to identify antigenic regions for designing an effective vaccine, which presents a substantial overhead and can be time-intensive [21]. This technique has been successfully implemented to develop a vaccine against various pathogens, for example, serogroup B Neisseria meningitides (MenB) [22].
In this study, an mRNA vaccine has been modeled, named MT. P495, using several bioinformatics tools targeting the phosphate-binding protein PstS1 of Mtb and also has been tested computationally for its ability to elicit immunogenic response and safety, predicted several types of T-cell and B-cell epitopes present within this antigen and their ability to generate an immune response within the host body. PstS1 protein is an immunodominant, TLR-2 agonist, inorganic phosphate up-taking lipoprotein found on the cell membrane surface of Mtb and also exhibits function as an adhesion molecule that facilitates binding with macrophage through mannose receptor (MR). This mRNA vaccine model thus serves as ready to test model in vivo by experimentalists and industries.
Materials and methods
A graphical depiction of the workflow is represented in Fig. 1A.

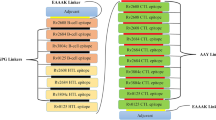
Graphical presentation of A steps involved in modeling an mRNA-based vaccine and B mRNA of the MT. P495 vaccine
Retrieval of protein sequence and 3D structure
A total of 3470 sequences of phosphate-binding protein PstS1 of Mtb were retrieved from the NCBI [23] database. The retrieved protein sequences had a length of 374 amino acids. The first 23 amino acids were the signal peptide and the rest of them encoded the PstS1 protein. Multiple sequence alignment (MSA) between the retrieved sequences was performed using the Clustal Omega [24] and the result was analyzed using the JalView [25]. The consensus sequence was retrieved from JalView and was used for sequence-based epitope prediction. The consensus sequence was 100% identical to the reference sequence of PstS1 retrieved from UniProt [26] (UniProtKB accession number P9WGU1). The X-ray crystal structure Mtb H37Rv PstS1 was obtained from RCSB PDB [27] (PDB ID-1PC3) for the prediction of structure-based discontinuous B-cell epitope.
Identification of cytotoxic T-lymphocyte (CTL) epitopes
To predict CD8+ CTL epitopes, NetCTL 1.2 Server [28] was used. The NetCTL server predicts CTL epitopes in a given protein sequence using the stabilized matrix base method [29]. The threshold level for prediction of a CTL epitope was set at 0.5 with a specificity of 0.94 along with a sensitivity of 0.89. Further analysis was performed on predicted epitopes with a higher combined score.
SMMPMBEC prediction method [30] of the T-cell epitope prediction resource of the Immune Epitope Database (IEDB) [31] was utilized to predict both types of MHC-I-binding alleles, occurring frequently as well as non-frequently. The half-maximal inhibitory concentration (IC50) threshold was set at 250 nM.
Helper T-lymphocyte (HTL) epitopes identification
The T-cell epitope prediction resource of the IEDB server was used to predict promiscuous CD4+ HTL epitopes. The NN-align 2.3 (NetMHCII 2.3) method [32] was employed in this study to predict MHC-II-binding alleles. Peptide length was set to be 15. For further analysis, predicted peptides having an IC50 value less than 50 nM were considered.
Evaluation of the selected epitopes
For the analysis of antigenic, allergenic and toxic properties of all predicted epitopes, VaxiJen v2.0 server [33], AllerTOP v.2.0 server [34] and ToxinPred server [35] were employed respectively. Based on an alignment-independent manner, VaxiJen predicts the antigenic probability of a given peptide [33]. In this study, the target organism was set to bacteria and the threshold was set to 0.5. For the categorization of allergenic and nonallergenic peptides, AllerTOP v.2.0 utilizes the k-nearest neighbours (kNN) method [34]. Employing both the dipeptide-based SVM algorithm and the MEME/MAST algorithm, the ToxinPred can predict the toxicity of the given peptide [35]. Using the IEDB conservancy analysis tool, the conservancy of the epitopes was determined [36] using a 100% sequence identity threshold.
Cytokine inducing ability of the HTL epitopes was subject to further assessment. IL4pred server [37] was used to predict the interleukin-4 (IL4) inducing ability of the selected peptides while Interleukin-10 (IL10) inducing ability was assessed using the IL10pred server [38]. Another property that was assessed of the HTL peptides was the interferon-γ (IFN-γ) inducing probability. IFNepitope server [39] was used in this study.
Estimation of population coverage
The population coverage of the selected CTL and HTL epitopes was evaluated. Employing the population coverage tool [40] of the IEDB server, population coverage of the selected epitopes and their corresponding MHC HLA-binding alleles was predicted.
Prediction of three-dimensional (3D) structure of the epitopes and HLA proteins
After confirmation of epitope antigenicity, non-allergenicity, non-toxicity, conservancy and the availability of alleles, the 3D structure of the selected 8 CTL peptides, as well as 3 HTL peptides, were predicted using the PEP-FOLD server at the Ressource Parisienne en Bioinformatique Structurale (RPBS) Mobyle Portal [41]. The five most probable structures were predicted by the server for each peptide sequence. For further investigation, the best-predicted structure with the lowest energy model was chosen.
In this study, HLA-C*12:03 and HLA-DRB1*01:01 were used for the validation of the binding of selected epitope and HLA molecules. The 3D structure of HLA-DRB1*01:01 was retrieved from RCSB PDB (PDB ID 4I5B). 3D structure of HLA-C*12:03 being unavailable in PDB, SWISS-MODEL server [42] that performs homology-based modeling, was used for the prediction of the 3D structure of this molecule. The sequence of the HLA-C*12:03 was retrieved from IPD-IMGT/HLA Database [43] (IMGT/HLA Accession No. HLA00455). The predicted structure was validated using PROCHECK [44], MolProbity [45], ProSA-web [46] and QMEAN [47].
Molecular docking of the selected epitopes
To confirm the interaction between the selected alleles and their respective epitopes, at the DINC 2.0 web-server [48], molecular docking was carried out following the protocol described elsewhere [49]. The box centre was set at 13.7, − 60.0, and − 20.2 Å in the X, Y, and Z axes, respectively, to predict the binding energy of HLA-C*12:03 with an epitope. Similarly, to predict the binding affinity of HLA-DRB1*01:01 with an epitope, the box centre, in this case, was set at 39.9, 47.5 and 93.0 Å in the X, Y, and Z axes, respectively. In both cases, box size was also set to automatic (based on ligand). Results from this server were further analyzed using BIOVIA Discovery Studio [50] and UCSF Chimera [51].
Molecular dynamics simulation
Following a procedure described elsewhere [49], MD simulations were carried out employing GROMACS (v2021) [52]. For the simulation process, CHARMM36 all-atom additive force field [53] was used. The protein was cleaned using UCSF Chimera dock prep functionality, and then missing residues were added by using the Dunbrack rotamer Library, employing an in-house python script. GROMACS pdb2gmx tool was used in order to add hydrogens to the protein. The GROMACS program was used to generate the protein topology for HLA and epitope. The system was contained within a cubic box of a simple point charge extended (SPC/E) water model [53], with a minimum distance of 1.0 nm between the wall and any component of the protein. The system was neutralized by adding an aqueous solution of Na+ (sodium) and Cl− (chloride) to a 0.15 M ionic strength.
For 100 picoseconds (ps), the system was equilibrated utilizing the NVT and NPT ensemble. For the next 100 ps under an isothermal ensemble, soft coupling with the Berendsen thermostat (NVT) [54] was used to progressively heat the minimized system to target temperatures. Position restrictions to the ligand were imposed before performing NVT simulations to prevent the ligand from drifting away from the protein during equilibration. Employing the LINCS algorithm [55], all of the bonds were restricted. At a temperature of 300 K, the NPT ensemble (constant pressure, and constant temperature simulation criteria) was performed, employing periodic boundary conditions (PBC).
The system was then coupled to a Parrinello–Rahman barostat [56] for an equilibration period of 100 ps at 1 bar of pressure. The Particle Mesh Ewald (PME) method was employed to process the electrostatic interactions. With a cutoff of 1.0 nm, the short-range van der Waals cutoff (rvdW) interactions were computed. Simulations were run for 100 ns. GROMACS was used to compute the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA) (using van der Waals Volumes and Radii) [57] and hydrogen bond (H-bond). In-house Python script with Matplotlib [58] and NumPy [59] library, as well as [R] (version 3.6.3) [60] Peptides library [61] were used to generate trajectory plots and figures.
Conformational B-cell epitopes identification
The conformational B-cell epitopes were identified by using the ElliPro [62] tool of the IEDB. ElliPro can identify both the linear and conformational B-cell epitopes based on the 3D structure of the given protein. In this study, the minimum PI (protrusion index) score was set to 0.5 and the maximum distance was set to 6 Å for the prediction of conformational B-cell epitopes.
Linear B-cell (LBL) epitopes identification
To predict LBL epitope, linear B-cell epitope prediction tool from IEDB was used. Epitopes were predicted using Emini surface accessibility prediction [63], BepiPred Linear Epitope Prediction 2.0 [64], Kolaskar and Tongaonkar antigenicity [65] and Karplus and Schulz flexibility prediction method [66], present at the IEDB server. Epitopes having a length between 10 and 40 amino acids were selected. Linear epitopes predicted by ElliPro were also selected. Antigenicity, allergenicity, toxicity and conservancy of the selected epitopes were assessed as described earlier.
Immune simulation of the epitopes
At the C-ImmSim server [67], in silico immune simulation was carried out for the characterization of the immune response profile of the selected peptides. Two injections of the target antigen were administered 4 weeks apart at 1 and 84 time-steps (wherein, the first dose is administered at time = 0 and each time-step is corresponding to 8 h in real life) with a dose of 1000 antigen proteins each, containing no LPS. The simulation was conducted for 5000 simulation steps. Host HLAs were selected according to their occurring frequency. Frequently occurring HLA alleles were selected to perform the study. Other simulation parameters were kept default.
Designing the vaccine mRNA construct
The open reading frame (ORF) of a conventional mRNA-based vaccine consists of five fundamental parts. The target antigen, also known as the gene of interest (GOI) is linked with an adjuvant by a linker. This construct is flanked by 5′ and 3′ untranslated regions (UTRs) and a terminal poly(A) tail. The 5′ end is capped by Cap1 (m7GpppNm) Cap. In this study, PstS1 was used as the GOI which exhibited high immunogenic activity in several studies [68,69,70]. As an adjuvant, 50S ribosomal protein L7/L12 (UniProtKB accession number P9WHE3) from Mtb was used while the signal peptide from the tissue plasminogen activator (tPA, UniProtKB accession number P00750) of Homo sapiens was also used.
Two peptide linkers were used to join the polypeptide chains. GGGGSEAAAKGGGGS linker was used to link the GOI and the adjuvant. Another peptide linker, AAY was used between the signal peptide and the adjuvant. In this study, the 5′ UTR from human β-globin gene (NCBI accession number NM_000518.5) along with the 3′ UTR from rabbit β-globin gene (GenBank [71] accession number V00882.1) were used to flank the construct. A 120-nucleotides (nts) long poly(A) tail was added to complete the vaccine construct (Fig. 1B). The construct was named MT. P495.
Optimizing codons and predicting secondary structure of the vaccine mRNA
For the vaccine mRNA to be efficiently translated by the host cells, codon optimization is important. Therefore, the codons of the final vaccine construct were optimized for efficient expression in human cells using several codon optimization tools; JCat [72], GeneArt Instant Designer by Thermofisher, GenSmart™ Codon optimization by GenScript (GS), Codon Optimization Tool by Integrated DNA Technologies (IDT). The quality of the optimized codons was analyzed Using Rare Codon Analysis tools by GS. This tool can predict the efficiency of the translation of the mRNA expressed as the codon adaptation index (CAI) value. Also, the presence of any tandem unusual codons can be detected, shown as codon frequency distribution (CFD). Based on these parameters, the best-optimized sequence was chosen for further assessment.
The secondary structure of the mRNA construct was predicted using the RNAfold tool of ViennaRNA Package 2.0 [73]. Both the minimum free energy (MFE) structure and the centroid secondary structure of the mRNA were obtained from this tool along with their MFE.
Physicochemical properties assessment of the vaccine peptide
For the assessment of physiochemical properties, several bioinformatics tools were used. tPA signal sequence was excluded as this segment will be cleaved by the protease, only the adjuvant and the target antigen were assessed for various properties. To predict the antigenicity of this peptide segment, VaxiJen v2.0 [33] and ANTIGENpro [74] were used. Allergenicity of the peptide was predicted using AllerTOP v.2.0 [34]. The toxicity of the peptide was predicted using ToxinPred [35]. Various physiochemical properties [i.e., theoretical isoelectric point (pI), instability index (II), aliphatic index (AI), and grand average of hydropathicity (GRAVY)] were predicted using ProtParam [75]. Adhesin probability was checked using Vaxign [111]. All the interacting amino acids of PstS1 with those antibodies were also identified in this study as conformational B-cell epitopes. MSA showed that most of the interacting residues were 100% conserved in all the isolates of Mtb. Twenty-two linear epitopes were also identified which were further analyzed for their antigenic, allergenic and toxicity profile. After analysis, eight epitopes were found to be safe and immunogenic. Among them, several were found to overlap with previously identified epitopes [112, 113] (Supplementary Table 4). These evidences suggest that PstS1 would be able to efficiently stimulate the B-cell epitope.
In this study, a theoretical mRNA vaccine, MT. P495 was delineated against Mtb. To construct the MT. P495 mRNA, PstS1 was used as the GOI. To generate a stronger immune response, an adjuvant was added with the GOI. Adjuvant plays an important role in improving humoral and cellular immune response [114, 115]. 50S ribosomal protein L7/L12 protein from Mtb was used as an adjuvant. 50S ribosomal protein L7/L12 itself is a TLR-4 agonist and has the potential to induce dendritic cell maturation [116]. The translation efficiency is also influenced by the length of the poly(A) tail. mRNAs with an A120 tail showed increased, prolonged expression of the protein and a poly(A) tail longer than A120 did not show any significant effect on the expression of the target protein [117]. UTRs from two different genes were used in this vaccine construct: 5′ UTR of the human β-globin gene and 3′ UTR of the rabbit β-globin gene. These UTRs are important for the efficient translation of the mRNA. The UTRs of the human β-globin gene can increase the efficiency of mRNA translation [118]. The 3′ UTR of rabbit β-globin gene can influence seroconversion, antibody titer and cytokine profiles [117, 119]. To cap the 5′ end, Cap1 (m7GpppNm) was used. The half-life of mRNA is also increased as a result of a synergistic effect of 5′ end cap and 3′ end poly(A) tail [120]. Another peptide chain, the signal peptide was obtained from the tPA of H. sapiens which can improve the immunogenicity of the vaccine [121]. Two different linkers were used to join different components of the vaccine construct. Linker molecules can join two polypeptide chains, the protein moieties [122]. They are also used for increasing the stability of the final mRNA product, the protein [122, 123]. The linker GGGGSEAAAKGGGGS can increase both the thermal as well as the pH stability of the fused protein [124]. Another linker, AAY is an in vivo cleavable linker that acts as a cleavage site of proteasomes [125]. The final length of the mRNA construct was 1856 nts.
The codons of the delineated mRNA were optimized for efficient translation and better expression in the host (Supplementary Fig. S9; Supplementary Table 5). The properties of the optimized sequence suggested that host cells will efficiently express the mRNA. No tandem unusual codons were identified which is also an indicator of efficient translation. For optimization of the codons, we integrated two different codon optimization tools. In vivo experiments also suggest that the combined use of these two tools can increase protein expression efficiency [126]. The optimized codons have been used for the prediction of the secondary structure of the mRNA. Predicted free energy and the structures indicated that the mRNA will be stable.
The physiochemical properties of the vaccine peptide were assessed to ensure the safety of the vaccine (Supplementary Table 6). The peptide was predicted to be antigenic, non-allergenic, and non-toxic. So, it will be safe for human usage. Since the protein will have a longer half-life in mammalian reticulocytes, it is likely to remain stable inside the host cells and will be bioavailable for a long time. After the confirmation of safety in the host, the structure of the vaccine peptide (Fig. 4) was predicted to confirm the binding ability of the vaccine peptide with TLRs and MR by the molecular docking study. Both of the ligand peptides (adjuvant-linked GOI and GOI alone) could bind with the receptors with a high binding affinity (Table 2). The binding affinity also indicated that the vaccine peptide will be able to bind with the receptors with a higher binding affinity than the GOI alone. It was also found that our vaccine peptide could bind with the receptors 100 times more tightly than GOI. The stability of the receptor–ligand complex was checked (Fig. 5). To perform this analysis, the vaccine–TLR4 complex was selected. The deformability graph and covariance map indicated that the complex will be stable.
Instead of using an in vivo method, IVT is a popular and preferred method for producing mRNA on a large-scale [127]. In this study, promoter and termination sequences specific to the T7 polymerase were used for the efficient synthesis of RNA in IVT. Maintaining the homogenous length of the poly(A) tail is a major problem of this method. Most of the time, the poly(A) tails vary in length if the circular plasmid is used. To solve this problem, the pJAZZ-OK® linear plasmid was utilized as a cloning vector [84]. The total length of the recombinant plasmid was 14,977 bp.
All of the results above, as well as previous research on humans as hosts, suggest that the proposed mRNA vaccine candidate, MT. P495, will probably elicit a strong immune response, specific against Mtb. In order to develop a viable Mtb vaccine in the future, this modeled mRNA is an excellent vaccine model that can be readily employed for laboratory testing, including in vitro as well as in vivo studies.
Data availability
The data supporting this article are available in both the article and its Online Supplementary Material.
References
Castro KG (1995) Tuberculosis as an opportunistic disease in persons infected with human immunodeficiency virus. Clin Infect Dis 21:66–71. https://doi.org/10.1093/clinids/21.Supplement_1.S66
Zhai W, Wu F, Zhang Y et al (2019) The immune escape mechanisms of Mycobacterium tuberculosis. Int J Mol Sci. https://doi.org/10.3390/ijms20020340
Heemskerk D, Caws M, Marais B, Farrar J (2015) Tuberculosis in adults and children. Springer, Cham
Shah P, Mistry J, Reche PA et al (2018) In silico design of Mycobacterium tuberculosis epitope ensemble vaccines. Mol Immunol 97:56–62. https://doi.org/10.1016/j.molimm.2018.03.007
Smith I (2003) Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev 16:463–496. https://doi.org/10.1128/CMR.16.3.463-496.2003
WHO (2021) Global tuberculosis report 2021: executive summary. World Health Organization, Geneva
Houben RMGJ, Dodd PJ (2016) The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med 13:1–13. https://doi.org/10.1371/journal.pmed.1002152
Whitlow E, Mustafa AS, Hanif SNM (2020) An overview of the development of new vaccines for tuberculosis. Vaccines 8:1–13. https://doi.org/10.3390/vaccines8040586
Wolf AJ, Desvignes L, Linas B et al (2008) Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med 205:105–115. https://doi.org/10.1084/jem.20071367
Doherty TM, Andersen P (2005) Vaccines for tuberculosis: novel concepts and recent progress. Clin Microbiol Rev 18:687–702. https://doi.org/10.1128/CMR.18.4.687-702.2005
Delogu G, Sali M, Fadda G (2013) The biology of Mycobacterium tuberculosis infection. Mediterr J Hematol Infect Dis. https://doi.org/10.4084/mjhid.2013.070
Schrager LK, Vekemens J, Drager N et al (2020) The status of tuberculosis vaccine development. Lancet Infect Dis 20:e28–e37. https://doi.org/10.1016/S1473-3099(19)30625-5
Orme IM (2015) Tuberculosis vaccine types and timings. Clin Vaccine Immunol 22:249–257. https://doi.org/10.1128/CVI.00718-14
Day CL, Tameris M, Mansoor N et al (2013) Induction and regulation of T-cell immunity by the novel tuberculosis vaccine M72/AS01 in South African adults. Am J Respir Crit Care Med 188:492–502. https://doi.org/10.1164/rccm.201208-1385OC
Pardi N, Hogan MJ, Porter FW, Weissman D (2018) mRNA vaccines—a new era in vaccinology. Nat Rev Drug Discov 17:261–279. https://doi.org/10.1038/nrd.2017.243
Pascolo S (2004) Messenger RNA-based vaccines. Expert Opin Biol Ther 4(8):1285–1294
Verbeke R, Lentacker I, De Smedt SC, Dewitte H (2019) Three decades of messenger RNA vaccine development. Nano Today 28:100766. https://doi.org/10.1016/j.nantod.2019.100766
Bian H (2003) The use of bioinformatics for identifying class II-restricted T-cell epitopes. Methods 29:299–309. https://doi.org/10.1016/S1046-2023(02)00352-3
Sirskyj D, Diaz-Mitoma F, Golshani A et al (2011) Innovative bioinformatic approaches for develo** peptide-based vaccines against hypervariable viruses. Immunol Cell Biol 89:81–89. https://doi.org/10.1038/icb.2010.65
Jabbar B, Rafique S, Salo-Ahen OMH et al (2018) Antigenic peptide prediction from E6 and E7 oncoproteins of HPV types 16 and 18 for therapeutic vaccine design using immunoinformatics and MD simulation analysis. Front Immunol. https://doi.org/10.3389/fimmu.2018.03000
Yang Z, Bogdan P, Nazarian S (2021) An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci Rep 11:3238. https://doi.org/10.1038/s41598-021-81749-9
Jain R, Singh S, Verma SK, Jain A (2019) Genome-wide prediction of potential vaccine candidates for Campylobacter jejuni using reverse vaccinology. Interdiscip Sci Comput Life Sci 11:337–347. https://doi.org/10.1007/s12539-017-0260-5
Wheeler DL, Barrett T, Benson DA et al (2007) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 35:5–12. https://doi.org/10.1093/nar/gkl1031
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. https://doi.org/10.1038/msb.2011.75
Waterhouse AM, Procter JB, Martin DMA et al (2009) JalView Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Bateman A (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. https://doi.org/10.1093/nar/gky1049
Burley SK, Bhikadiya C, Bi C et al (2021) RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 49:D437–D451. https://doi.org/10.1093/nar/gkaa1038
Larsen MV, Lundegaard C, Lamberth K et al (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform 8:1–12. https://doi.org/10.1186/1471-2105-8-424
Peters B, Sette A (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform 6:1–9. https://doi.org/10.1186/1471-2105-6-132
Kim Y, Sidney J, Pinilla C et al (2009) Derivation of an amino acid similarity matrix for peptide:MHC binding and its application as a Bayesian prior. BMC Bioinform 10:394. https://doi.org/10.1186/1471-2105-10-394
Vita R, Mahajan S, Overton JA et al (2019) The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 47:D339–D343. https://doi.org/10.1093/nar/gky1006
Nielsen M, Lund O (2009) NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform 10:296. https://doi.org/10.1186/1471-2105-10-296
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform 8:1–7. https://doi.org/10.1186/1471-2105-8-4
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP vol 2—a server for in silico prediction of allergens. J Mol Model. https://doi.org/10.1007/s00894-014-2278-5
Gupta S, Kapoor P, Chaudhary K et al (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. https://doi.org/10.1371/journal.pone.0073957
Bui HH, Sidney J, Li W et al (2007) Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform 8:1–6. https://doi.org/10.1186/1471-2105-8-361
Dhanda SK, Gupta S, Vir P, Raghava GP (2013) Prediction of IL4 inducing peptides. Clin Dev Immunol 2013:263952. https://doi.org/10.1155/2013/263952
Nagpal G, Usmani SS, Dhanda SK et al (2017) Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep 7:1–10. https://doi.org/10.1038/srep42851
Dhanda SK, Vir P, Raghava GPS (2013) Designing of interferon-gamma inducing MHC class-II binders. Biol Direct 8:1–15. https://doi.org/10.1186/1745-6150-8-30
Bui HH, Sidney J, Dinh K et al (2006) Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform 7:1–5. https://doi.org/10.1186/1471-2105-7-153
Maupetit J, Derreumaux P, Tufféry P (2009) A fast method for large-scale De Novo peptide and miniprotein structure prediction. J Comput Chem. https://doi.org/10.1002/jcc.21365
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. https://doi.org/10.1093/nar/gky427
Robinson J, Barker DJ, Georgiou X et al (2020) IPD-IMGT/HLA database. Nucleic Acids Res 48:D948–D955. https://doi.org/10.1093/nar/gkz950
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. https://doi.org/10.1107/s0021889892009944
Williams CJ, Headd JJ, Moriarty NW et al (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27:293–315. https://doi.org/10.1002/pro.3330
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:407–410. https://doi.org/10.1093/nar/gkm290
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350. https://doi.org/10.1093/bioinformatics/btq662
Antunes DA, Moll M, Devaurs D et al (2017) DINC 2.0: a new protein–peptide docking webserver using an incremental approach. Cancer Res 77:e55–e57. https://doi.org/10.1158/0008-5472.CAN-17-0511
Shahrear S, Islam ABMMK (2022) Immunoinformatics guided modeling of CCHF_GN728, an mRNA-based universal vaccine against Crimean-Congo hemorrhagic fever virus. Comput Biol Med 140:105098. https://doi.org/10.1016/j.compbiomed.2021.105098
Dassault Systmes (2017) BIOVIA Discovery Studio Dassault Systmes BIOVIA, discovery studio modeling environment, Release 2017. Dassault Systmes
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91:43–56. https://doi.org/10.1016/0010-4655(95)00042-E
Berendsen HJC, Grigera JR, Straatsma TP (1987) The missing term in effective pair potentials. J Phys Chem 91:6269–6271. https://doi.org/10.1021/j100308a038
Berendsen HJC, Postma JPM, van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. https://doi.org/10.1063/1.448118
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. https://doi.org/10.1002/(SICI)1096-987X(199709)18:12%3c1463::AID-JCC4%3e3.0.CO;2-H
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. https://doi.org/10.1063/1.328693
Bondi A (1964) van der Waals volumes and radii. J Phys Chem 68:441–451. https://doi.org/10.1021/j100785a001
Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9:90–95. https://doi.org/10.1109/MCSE.2007.55
Harris CR, Millman KJ, van der Walt SJ et al (2020) Array programming with NumPy. Nature 585:357–362. https://doi.org/10.1038/s41586-020-2649-2
R Core Team (2021) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. Accessed 02 May 2022
Osorio D, Rondón-Villarreal P, Torres R (2015) Peptides: a package for data mining of antimicrobial peptides. R J 7:4. https://doi.org/10.32614/RJ-2015-001
Ponomarenko J, Bui HH, Li W et al (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform 9:1–8. https://doi.org/10.1186/1471-2105-9-514
Emini EA, Hughes JV, Perlow DS, Boger J (1985) Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol 55:836–839. https://doi.org/10.1128/jvi.55.3.836-839.1985
Jespersen MC, Peters B, Nielsen M, Marcatili P (2017) BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res 45:W24–W29. https://doi.org/10.1093/nar/gkx346
Kolaskar AS, Tongaonkar PC (1990) A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 276:172–174. https://doi.org/10.1016/0014-5793(90)80535-Q
Karplus PA, Schulz GE (1985) Prediction of chain flexibility in proteins—a tool for the selection of peptide antigens. Naturwissenschaften 72:212–213. https://doi.org/10.1007/BF01195768
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE. https://doi.org/10.1371/journal.pone.0009862
Gottschalk N, Lang S, Kimmig R et al (2012) Monocytes and the 38 kDa-antigen of Mycobacterium tuberculosis modulate natural killer cell activity and their cytolysis directed against ovarian cancer cell lines. BMC Cancer 12:1. https://doi.org/10.1186/1471-2407-12-451
Young DB, Garbe TR (1991) Lipoprotein antigens of Mycobacterium tuberculosis. Res Microbiol 142:55–65. https://doi.org/10.1016/0923-2508(91)90097-T
Liu H, Jiang Y, Dou X et al (2013) PstS1 polymorphisms of Mycobacterium tuberculosis strains may reflect ongoing immune evasion. Tuberculosis 93:475–481. https://doi.org/10.1016/j.tube.2013.05.003
Agarwala R, Barrett T, Beck J et al (2018) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 46:D8–D13. https://doi.org/10.1093/nar/gkx1095
Grote A, Hiller K, Scheer M et al (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:526–531. https://doi.org/10.1093/nar/gki376
Gruber AR, Lorenz R, Bernhart SH et al (2008) The Vienna RNA websuite. Nucleic Acids Res 36:70–74. https://doi.org/10.1093/nar/gkn188
Magnan CN, Zeller M, Kayala MA et al (2010) High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 26:2936–2943. https://doi.org/10.1093/bioinformatics/btq551
Gasteiger E, Hoogland C, Gattiker A et al (2005) Protein identification and analysis tools on the ExPASy server. In: The proteomics protocols handbook. Humana Press, Totowa, pp 571–607
He Y, **ang Z, Mobley HLT (2010) Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol. https://doi.org/10.1155/2010/297505
Buchan DWA, Jones DT (2019) The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 47:W402–W407. https://doi.org/10.1093/nar/gkz297
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. https://doi.org/10.1006/jmbi.1999.3091
Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:526–531. https://doi.org/10.1093/nar/gkh468
Kozakov D, Hall DR, **a B et al (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12:255–278. https://doi.org/10.1038/nprot.2016.169
Xue LC, Rodrigues JP, Kastritis PL et al (2016) PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 32:3676–3678. https://doi.org/10.1093/bioinformatics/btw514
López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P (2014) iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res 42:271–276. https://doi.org/10.1093/nar/gku339
Jackson NAC, Kester KE, Casimiro D et al (2020) The promise of mRNA vaccines: a biotech and industrial perspective. NPJ Vaccines 5:11. https://doi.org/10.1038/s41541-020-0159-8
Grier AE, Burleigh S, Sahni J et al (2016) pEVL: a linear plasmid for generating mRNA IVT templates with extended encoded poly(A) sequences. Mol Ther Nucleic Acids 5:e306. https://doi.org/10.1038/mtna.2016.21
Courel M, Clément Y, Bossevain C et al (2019) GC content shapes mRNA storage and decay in human cells. eLife. https://doi.org/10.7554/eLife.49708
Crampin AC, Glynn JR, Fine PEM (2009) What has Karonga taught us? Tuberculosis studied over three decades. Int J Tuberc Lung Dis 13:153–164
Pollard C, De Koker S, Saelens X et al (2013) Challenges and advances towards the rational design of mRNA vaccines. Trends Mol Med 19:705–713. https://doi.org/10.1016/j.molmed.2013.09.002
Iavarone C, O’Hagan DT, Yu D et al (2017) Mechanism of action of mRNA-based vaccines. Expert Rev Vaccines 16:871–881. https://doi.org/10.1080/14760584.2017.1355245
Tait DR, Hatherill M, Van Der Meeren O et al (2019) Final analysis of a trial of M72/AS01 E vaccine to prevent tuberculosis. N Engl J Med 381:2429–2439. https://doi.org/10.1056/nejmoa1909953
Okoye AA, Picker LJ (2013) CD4+ T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev 254:54–64. https://doi.org/10.1111/imr.12066
Baden LR, El Sahly HM, Essink B et al (2021) Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med 384:403–416. https://doi.org/10.1056/nejmoa2035389
Polack FP, Thomas SJ, Kitchin N et al (2020) Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med 383:2603–2615. https://doi.org/10.1056/nejmoa2034577
Voysey M, Clemens SAC, Madhi SA et al (2021) Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 397:99–111. https://doi.org/10.1016/S0140-6736(20)32661-1
Walsh EE, Frenck RW, Falsey AR et al (2020) Safety and immunogenicity of two RNA-based COVID-19 vaccine candidates. N Engl J Med 383:2439–2450. https://doi.org/10.1056/nejmoa2027906
Sahin U, Muik A, Derhovanessian E et al (2020) COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 586:594–599. https://doi.org/10.1038/s41586-020-2814-7
Mulligan MJ, Lyke KE, Kitchin N et al (2020) Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586:589–593. https://doi.org/10.1038/s41586-020-2639-4
**ao T, Jiang Y, Li G et al (2019) Polymorphism of MPT64 and PstS1 in Mycobacterium tuberculosis is not likely to affect relative immune reaction in human. Medicine (US) 98:1–6. https://doi.org/10.1097/MD.0000000000018073
Weber CA, Mehta PJ, Ardito M et al (2009) T cell epitope: Friend or Foe? Immunogenicity of biologics in context. Adv Drug Deliv Rev 61:965–976. https://doi.org/10.1016/j.addr.2009.07.001
Peters B, Bulik S, Tampe R et al (2003) Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol 171:1741–1749. https://doi.org/10.4049/jimmunol.171.4.1741
Arango Duque G, Descoteaux A (2014) Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. https://doi.org/10.3389/fimmu.2014.00491
Vordermeier H-M, Harris DP, Friscia G et al (1992) T cell repertoire in tuberculosis: selective anergy to an immunodominant epitope of the 38-kDa antigen in patients with active disease. Eur J Immunol 22:2631–2637. https://doi.org/10.1002/eji.1830221024
Wilkinson KA, Vordermeier MH, Kajtár J et al (1997) Modulation of peptide specific T cell responses by non-native flanking regions. Mol Immunol 34:1237–1246. https://doi.org/10.1016/S0161-5890(98)00009-1
Wilkinson RJ, Vordermeier HM, Wilkinson KA et al (1998) Peptide-specific T cell response to Mycobacterium tuberculosis: clinical spectrum, compartmentalization, and effect of chemotherapy. J Infect Dis 178:760–768. https://doi.org/10.1086/515336
Wilkinson KA, Vordermeier H, Wilkinson RJ et al (1998) Synthesis and in vitro T-cell immunogenicity of conjugates with dual specificity: attachment of epitope peptides of 16 and 38 kDa proteins from Mycobacterium tuberculosis to branched polypeptide. Bioconjug Chem 9:539–547. https://doi.org/10.1021/bc970159+
Venkataprasad N (1999) Induction of cellular immunity to a mycobacterial antigen adsorbed on lamellar particles of lactide polymers. Vaccine 17:1814–1819. https://doi.org/10.1016/S0264-410X(98)00372-7
Liu J, Chen X, Wang J et al (2021) Prediction and identification of CD4+ T cell epitope for the protective antigens of Mycobacterium tuberculosis. Medicine (Baltim) 100:e24619. https://doi.org/10.1097/MD.0000000000024619
da Silva BCM, Grassi MFR, Coutinho R et al (2014) Mycobacterium tuberculosis epitope-specific interferon-g production in healthy Brazilians reactive and non-reactive to tuberculin skin test. Mem Inst Oswaldo Cruz 109:999–1004. https://doi.org/10.1590/0074-0276140193
Hammond AS, Klein MR, Corrah T et al (2005) Mycobacterium tuberculosis genome-wide screen exposes multiple CD8+ T cell epitopes. Clin Exp Immunol 140:109–116. https://doi.org/10.1111/j.1365-2249.2005.02751.x
Shams H, Barnes PF, Weis SE et al (2003) Human CD8+ T cells recognize epitopes of the 28-kDa hemolysin and the 38-kDa antigen of Mycobacterium tuberculosis. J Leukoc Biol 74:1008–1014. https://doi.org/10.1189/jlb.0403138
Cho S, Mehra V, Thoma-Uszynski S et al (2000) Antimicrobial activity of MHC class I-restricted CD8+ T cells in human tuberculosis. Proc Natl Acad Sci USA 97:12210–12215. https://doi.org/10.1073/pnas.210391497
Watson A, Li H, Ma B et al (2021) Human antibodies targeting a Mycobacterium transporter protein mediate protection against tuberculosis. Nat Commun. https://doi.org/10.1038/s41467-021-20930-0
Gaseitsiwe S, Valentini D, Mahdavifar S et al (2008) Pattern recognition in pulmonary tuberculosis defined by high content peptide microarray chip analysis representing 61 proteins from M. tuberculosis. PLoS ONE 3:e3840. https://doi.org/10.1371/journal.pone.0003840
López-Vidal Y, de León-Rosales SP, Castañón-Arreola M et al (2004) Response of IFN-γ and IgG to ESAT-6 and 38 kDa recombinant proteins and their peptides from Mycobacterium tuberculosis in tuberculosis patients and asymptomatic household contacts may indicate possible early-stage infection in the latter. Arch Med Res 35:308–317. https://doi.org/10.1016/j.arcmed.2004.04.008
Sarkar I, Garg R, van Drunen Littel-van den Hurk S (2019) Selection of adjuvants for vaccines targeting specific pathogens. Expert Rev Vaccines 18:505–521. https://doi.org/10.1080/14760584.2019.1604231
Coccia M, Collignon C, Hervé C et al (2017) Cellular and molecular synergy in AS01-adjuvanted vaccines results in an early IFNγ response promoting vaccine immunogenicity. NPJ Vaccines. https://doi.org/10.1038/s41541-017-0027-3
Lee SJ, Shin SJ, Lee MH et al (2014) A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE 9:1–11. https://doi.org/10.1371/journal.pone.0104351
Holtkamp S, Kreiter S, Selmi A et al (2006) Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108:4009–4017. https://doi.org/10.1182/blood-2006-04-015024
Zhuang X, Qi Y, Wang M et al (2020) mRNA vaccines encoding the HA protein of influenza a H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccines 8:1–17. https://doi.org/10.3390/vaccines8010123
Zinckgraf JW, Silbart LK (2003) Modulating gene expression using DNA vaccines with different 3′-UTRs influences antibody titer, seroconversion and cytokine profiles. Vaccine 21:1640–1649. https://doi.org/10.1016/S0264-410X(02)00740-5
Gallie DR (1991) The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev 5:2108–2116. https://doi.org/10.1101/gad.5.11.2108
Kou Y, Xu Y, Zhao Z et al (2017) Tissue plasminogen activator (tPA) signal sequence enhances immunogenicity of MVA-based vaccine against tuberculosis. Immunol Lett 190:51–57. https://doi.org/10.1016/j.imlet.2017.07.007
Chen X, Zaro JL, Shen WC (2013) Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65:1357–1369. https://doi.org/10.1016/j.addr.2012.09.039
Arai R, Wriggers W, Nishikawa Y et al (2004) Conformations of variably linked chimeric proteins evaluated by synchrotron X-ray small-angle scattering. Proteins Struct Funct Genet 57:829–838. https://doi.org/10.1002/prot.20244
Chen H, Chen Z, Wu B et al (2017) Influences of various peptide linkers on the Thermotoga maritima MSB8 nitrilase displayed on the spore surface of Bacillus subtilis. J Mol Microbiol Biotechnol 27:64–71. https://doi.org/10.1159/000454813
Agallou M, Margaroni M, Kotsakis SD, Karagouni E (2020) A canine-directed chimeric multi-epitope vaccine induced protective immune responses in BALB/C mice infected with Leishmania infantum. Vaccines 8:1–35. https://doi.org/10.3390/vaccines8030350
Koblan LW, Doman JL, Wilson C et al (2018) Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36:843–846. https://doi.org/10.1038/nbt.4172
Kanwal F, Chen T, Zhang Y et al (2018) Large-scale in vitro transcription, RNA purification and chemical probing analysis. Cell Physiol Biochem 48:1915–1927. https://doi.org/10.1159/000492512
Acknowledgements
The authors acknowledge support from Biomedical Research Foundation (BMRF, DU). Authors also acknowledge High Performance Computing Facility support from Centre for Bioinformatics Learning Advancement and Systematics Training (cBLAST), University of Dhaka.
Funding
The project was not funded by any internal or external organization.
Author information
Authors and Affiliations
Contributions
The project was designed by ABMMKI. The data was collected by SS. The analysis was performed by SS and ABMMKI. The draft was written by SS and ABMMKI. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declared that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shahrear, S., Islam, A.B.M.M.K. Modeling of MT. P495, an mRNA-based vaccine against the phosphate-binding protein PstS1 of Mycobacterium tuberculosis. Mol Divers 27, 1613–1632 (2023). https://doi.org/10.1007/s11030-022-10515-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10515-4