
Abstract
Epithelial-to-mesenchymal transition (EMT), which plays an essential role in development, tissue repair and fibrosis, and cancer progression, is a reversible cellular program that converts epithelial cells to mesenchymal cell states characterized by motility-invasive properties. The mostly signaling pathways that initiated and controlled the EMT program are regulated by a solitary, non-motile organelle named primary cilium. Acting as a signaling nexus, primary cilium dynamically concentrates signaling molecules to respond to extracellular cues. Recent research has provided direct evidence of connection between EMT and primary ciliogenesis in multiple contexts, but the mechanistic understanding of this relationship is complicated and still undergoing. In this review, we describe the current knowledge about the ciliary signaling pathways involved in EMT and list the direct evidence that shows the link between them, trying to figure out the intricate relationship between EMT and primary ciliogenesis, which may aid the future development of primary cilium as a novel therapeutic approach targeted to EMT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The epithelial-to-mesenchymal transition (EMT) is a reversible cellular program that converts epithelial cells to mesenchymal cell states characterized by the motility-invasive properties with altered expression of the epithelial marker E-cadherin and the mesenchymal marker vimentin [1]. The classified EMT programs as EMT-type 1, 2 and 3 by biological context operate vital roles for gastrulation, neural crest delamination during embryonic development [2], tissue repair and fibrosis [3], cancer progression [4]. Although it has been viewed as a binary process between these two extremes, recent studies indicated that EMT is a continuous dynamic transition process from epithelial to completely mesenchymal states. It passes through intermediate hybrid states that express different levels of epithelial and mesenchymal markers and exhibits intermediate morphological, transcriptional and epigenetic features [5, 6].
EMT is mainly regulated by a core set of EMT-activating transcription factors (EMT-TFs), including SNAIL (SNAI1) and SLUG (SNAI2), the basic helix–loop–helix factors TWIST1 (TWIST) and TWIST2 and the zinc finger E-Box binding homeobox factors ZEB1 and ZEB2 to repress epithelial genes like the E-cadherin and upregulate the expression of mesenchymal genes like vimentin [7]. Meanwhile, multiple signaling pathways such as transforming growth factor-β(TGFβ), WNT, NOTCH, and phosphatidylinositol 3-kinase-AKT kinase (PI3K-AKT) cooperate in the initiation and progression of EMT by ultimately inducing the expression of various EMT-TFs during normal development, wound healing and carcinoma progression [8].
The primary cilium is a solitary, hair-like organelle elongating from the cell surface into extracellular environments in almost all human cell types [9]. As a functional antenna, besides its role to receive information from the environment and locally transduce it into a cellular response, researchers also revealed that primary cilium could transmit signals through released extracellular vesicles (EVs) [10, 11]. Thus a suite of soluble ligands such as Sonic hedgehog (SHH), Receptor Tyrosine Kinases (RTK), WNT, G-protein-coupled receptors (GPCR) and NOTCH were dynamically received and transduced in part or sum by primary cilium [12], which activates intracellular or extracellular signaling pathways to regulate cell polarization, differentiation, proliferation, specific immune cell functions and metabolism [13, 14]. The dysfunction of primary cilium was also proposed as a prerequisite step of cancer development and stem cell identity [15, 16]. Although the study on role of primary cilium in cancer is still undergoing, it is well accepted that the primary cilium appear to have a dual function in different cancers or even same cancer by either promoting or blocking tumorigenesis depending on the nature of the oncogenic initiating event[17].
Emerging evidence indicated that primary cilium mutation interrupted EMT in multi organs and tissues, including epicardial tissue, kidney epithelial cells, pancreatic β-cells and the retinal pigment epithelium (RPE) [18,19,20,21,22]. Recent research also revealed the connection between EMT and primary ciliogenesis in the context of cancer such as glioblastoma(GBM) and triple-negative breast cancer (TNBC) [23, 24], bladder cancer [25], proposing the relevance of primary cilium as a potential target in cancer EMT. Nevertheless, the mechanism is still complicated. The focus of this review is to described the current knowledge about the influence of ciliary signaling pathways for EMT and try to figure out the intricate relationship between EMT and primary ciliogenesis, which maybe aid the future development of primary cilium as a novel therapeutic approach targeted to EMT.
Ciliary signaling pathway involved in EMT
EMT can be induced by several intracellular signaling pathways that are known to induce the expression of various EMT-TFs [8, 26],in which the primary cilium function as a mediator by detecting signals from the extracellular environment and displaying specific receptors required for signal interception and the downstream molecular effectors [27]. In this section, we focus on the ciliary signaling pathways functioned in EMT including TGFβ, HH, WNTs, NOCTH and RTK.
Primary cilium-based TGFβ signaling
The superfamily of TGFβ comprises more than 30 different ligands of TGFβ/activin/Nodal and bone morphogenetic protein (BMP) subfamilies to activate serine/threonine kinases receptors of types I and II (TGF-βRI/II and BMP-RI/II, respectively) in paracrine or autocrine manners [28], which function in a multitude of cellular processes during development and in the maintenance of tissue homeostasis in the adult.
Increasing evidence demonstrate the remarkable role for the primary cilium in regulating TGFβ signaling. The first report of active TGFβ signaling in the primary cilium was based on studies using cultured mice and human fibroblasts. The localization of TGFβ receptors were identified in the tip of the cilium using immunofluorescence microscopy, when activated the TGFβ receptors migrated to the ciliary base to phosphorylate SMAD2/3 and ERK1/2. TGFβ signaling were reduced at stunted primary cilium in Tg737orpk fibroblasts, indicating that the primary cilium regulate the output of canonical and non-canonical TGFβ signaling to control varied cellular responses [29]. Further studies supported the idea that primary cilium is vital for balancing the output of TGF-β/BMP signaling to control varied cellular responses such as mice heart development [30], migration of human bone mesenchymal stem cells [31] and migration and tumor metastasis in mammary cancer cells [32]. TGFβ-1 signaling and SMAD3 activation were impaired in ciliated fibroblasts which are enriched in areas of myocardial injury upon primary cilium being removed, extracellular matrix protein levels and contractile function were also impaired, suggesting a pivotal role of primary cilium in disease-related pathological cardiac remodeling via regulating TGF-β1/SMAD3 axis [33]. Meanwhile, IFT80 deletion down-regulated the TGF-β signaling pathway by inhibiting the expression of TGF-βI, TGF-βR, and phosphorylation of Smad2/3 in the fracture healing [34]. Moreover, other proteins that regulate receptor transport and feedback inhibition mechanisms are enriched in cilia-centrosome axis, which underlined the importance of cilium in coordinating TGFβ signaling. For instance, loss of CEP128, a sub distal appendage protein which recruited RAB11 [35] that is responsible for endosomal recycling of TGFβ receptors to cilium, impaired phosphorylation of SMADs leading to defective organ development in zebrafish [36] and in male fertility [37]. Interestingly, TGF-β treatment resulted in suppresses average length of cilia by upregulating histone deacetylase (HDAC) activity [38] and regulating Ift88 gene expression at least in part via posttranscriptional manner [39].Conversely, SMAD7, the feedback inhibitor of TGFβ signaling, which localize to ciliary base and restrain excessive signaling from primary cilium [29] had been proposed to limit ARL6-mediated ciliary localization of TGFβ receptors and suppress tumor cell migration and invasion by restricting cross-talk between TGFβ receptors and SMO in HH signaling [32].
Primary cilium-based Hedgehog signaling
Hedgehog (HH) signaling is an intricate, highly conserved evolutionary pathway that plays an instructional role during embryonic development, stem cell biology and tissue homeostasis [40].
Primary cilium play a vital role in HH signal transduction was well established. At first, researchers found that defects in intraflagellar transport (IFT) disrupted sonic hedgehog (SHH) signaling based on the neural tube patterning of mice embryos [41]. Further evidence indicated that dysfunction of primary cilium might disturb HH signaling in different models. For example, The ADP ribosylation factor-like GTPase 13B (ARL13B) is extensively used as a primary cilium marker and was found to mediate ciliary entry of SMO, mice lacking ARL13B had abnormal SHH signaling [42, 43]. It is well accepted that in vertebrates, the core HH machinery components, including Patched 1 (PTCH1), GPR161 and Smoothened (SMO), suppressor of fused (SUFU) and the glioma-associated oncogene (GLI) transcription factors which dynamically localized to primary cilium throughout the cell cycle [44, 45].To be more specific, when signaling is activated, in addition to PTCH1 removal, SMO accumulates in the ciliary membrane [46] and changes the processing of Gli, giving rise to the Gli variant GliA, which then leaves cilium and enters nucleus to activate downstream genes that change the differentiation program [47]. At the same time, GPR161, which was identified as a negative regulator of SHH signaling [48], was removed from primary cilium [49], in a β-arrestin-dependent manner [50].Therefore, canonical HH signaling is strictly dependent on intact primary cilium and IFT [40]. Prominin-1 (Prom1, also known as CD133),which involve in the maintenance of ciliary structure and function [51], mediate the transduction of Glis2 from primary cilium to nucleus and activation of the direct downstream target STAT3 in mouse incisor cervical loop epithelium-associated stem cells [52].The role of primary cilium in regulating non-canonical HH pathways is still little known, but ciliary protein IFT80 can inhibits HH non-canonical signaling via HH–Smo–Gai–RhoA–stress fiber signaling while promoting HH canonical signaling in mouse osteoblast differentiation [53].
Remarkably, studies show that primary cilium can either boost or inhibit tumorigenesis depended on the underlying carcinogenic factors in cancer types [54, 55]. Ciliary ablation strongly inhibited BCC-like tumors induced by an activated form of Smoothened. In contrast, removal of cilia accelerated tumors induced by activated Gli2, a transcriptional effector of Hh signaling [54].Conversely, knocked down of Stk11, also known as Lkb1,which was identified as a HH pathway gene using genome-wide RNA interference (RNAi) screen resulted in increased Gli3R abundance and cilia disassembly [56], suggesting that HH pathway may also regulate primary ciliogenesis and maintenance in a feedback loop.
Primary cilium-based WNT signaling
The WNT signaling pathway is a critical molecular rheostat to regulate development and adult tissue homeostasis, and its dysregulation has been found in many cancer types. For example, alterations in the WNT signaling pathway are a near-universal feature of colorectal cancer driven by truncating mutations in adenomatosis polyposis coli (APC) [57]. WNT signaling can induce β-catenin-dependent (canonical) and β-catenin-independent (non-canonical) signaling. The latter includes calcium signaling or the planar cell polarity (PCP) pathway [58].
Several core WNT pathway components were found to have ciliated localization [59, 60] and INVERSIN inhibits the canonical WNT pathway by targeting cytoplasmic disheveled (Dsh or Dvl1) for degradation first link WNT signaling with primary cilium [61]. Some studies supported that primary cilium (or cilium-associated proteins) promote tumors by restraining WNT signaling activity from regulating proliferation and differentiation. For instance, some researchers found that downregulation of cilium proteins including KIF3A, IFT88 and OFD1 in mice embryos, primary fibroblasts and embryonic stem cells leads to accumulating β-catenin, which subsequently increases the transcription of WNT target genes [62]. Further research revealed a spatial mechanism that the primary cilium diverted Jouberin (JBN), a ciliopathy protein and β-catenin-positive regulator, by facilitating its nuclear localization away from the nucleus and thereby negatively affecting WNT signaling [63]. Studies [64, 65] have shown that deletion of BBSome (a complex containing eight Bardet Biedl syndrome proteins: BBS 1, 2, 4, 5, 7, 8, 9 and 18) influenced stabilization and post-translational modification of β-catenin by perturbing proteasomal degradation and regulating histone deacetylase 6 (HDAC6), an enzyme that deacetylates β-catenin at lysine 49 and inhibit β-catenin phosphorylation at serine 45 [66], to alter the regulation of downstream WNT targets and establish the PCP mutant phenotypes, including open eyelids and disorganized stereocilium in mice model [67] and left–right asymmetry in zebrafish [68]. Meanwhile, increased phosphorylation of transcriptionally active serine 552 on β-catenin was observed in BBS8 knockdown RPE cells, offering proof for BBS8 suppressing canonical WNT signaling [69, 70]. Further research revealed that deletion of BBS8 disrupted asymmetric accumulation of the core PCP protein Vangl2 in cochlear cells, suggesting a role for BBS8 possibly upstream of core PCP asymmetry. Loss of INVERSIN and OFD1 led to PCP-regulated convergent extension defects in vertebrates also supported the involvement of cilium in non-canonical WNT signaling [61, 71]. Besides that, the deletion of BBSome had increased the release of small EVs (smEVs) loaded with WNT-related molecules and smEVs derived from ciliopathy patient renal tissues dampened the WNT response in target cells, in contrast with control tissues [72]. Although the involvement of primary cilium in WNT signaling was still contentious and undergoing, these data suggest that primary cilium is required for canonical or non-canonical WNT signaling.
Primary cilium-based NOTCH signaling
The NOTCH signaling is an evolutionarily conserved pathway crucial in regulating a diverse array of cell fate decisions during lineage commitment, differentiation, cell cycle progression, maintenance and self-renewal of stem cells [73]. Activation of the NOTCH signaling depends on the combination of Delta-like and Jagged families NOTCH ligands with the receptors of NOTCH1-4 [74].
A role for the primary cilium in NOTCH signaling was first identified in 2011 when the knockdown of IFT proteins in both cultured keratinocytes and embryonic epidermis cells led to significant NOTCH defects, while differentiation defects were cell-autonomous and rescued by activated NOTCH [75]. Subsequent research by the same group showed that the small GTPase (ARF4)-dependent polarized exocytosis acts through the basal body-ciliary complex to regulate NOTCH signaling during epidermal differentiation spatially [76]. Loss of primary cilium in corneal epithelial diminished NOTCH activation to reduce cell proliferation, accompanied by NOCTH1 and NOTCH2 receptors which were normally expressed and nuclear NOTCH1 Intracellular Domain (NICD1) was severely reduced [77]. Hemodynamic alteration is perceived by endocardial cells through primary cilium that mediates the upregulation of hemodynamic responsive factors klf2a and klf2b. The increased klf2 gene expression activates endocardial NOTCH signaling to promote ventricle regeneration [78]. Liu et al. reported that primary cilium regulates hematopoietic stem and progenitor cell specification through NOTCH signaling in zebrafish [79]. A study by Leitch et al. showed that the absence of BBSome subunits resulted in the upregulation of NOTCH signaling in transgenic zebrafish NOTCH receptor cell lines and human cultured cells. The reduction of BBS1 or BBS4 diminished the recycling of NOTCH from early endosomes [80]. Although the exact regulatory process and the underlying mechanisms remain incomplete, these evidence depicted that primary cilium regulates NOTCH signaling by modulating the spatial localization of NOTCH signaling intermediates.
Primary cilium-based RTK signaling
RTK signaling consists of 58 members, which plays an essential role in various cellular processes including growth, motility, differentiation and metabolism in humans [81]. Among them, epidermal growth factor receptors (EGFRs), platelet-derived growth factor receptors (PDGFRs), insulin-like growth factor receptors (IGFRs), fibroblast growth factor receptors (FGFRs) and Tropomyosin receptor kinase B(TRKB) were linked to primary cilium by ciliary localization and associated functions [82]. PDGFRα-mediated signaling depends upon its ciliary localization in quiescent fibroblasts and fibroblasts derived from Tg737orpk mutants, which failed to form normal cilium and upregulate the level of PDGFRα. Thus, the Mek1/2-Erk1/2 pathway could not be activated [83]. Further research from the same team demonstrated that in coordination with the cytoskeletal reorganization, the fibroblast primary cilium function via ciliary PDGFRα signaling to monitor directional cell migration during wound healing [84]. The depletion of cilium disassembly complex components is sufficient to induce ciliogenesis in a subset of glioma stem cells (GSCs). The process occurs via relocating PDGFRα to the newly induced cilium and reintroducing cilium switches GSCs from self-renewal to differentiation. This can prevent the infiltration of GSCs into the brain, suggesting cilium induction as an attractive strategy to intervene in GSCs proliferation via PDGFRα signaling [85]. Less activated AKT, which was shown to be activated at the ciliary base in a PDGF-AA-dependent manner [86], led to impaired migration response in BBS1M390R/M390R MEF cells [87]. Mechanisms that regulate ciliary targeting of PDGFRα and balance the output of PDGF-AA-mediated signaling have been revealed. A recent study revealed that IFT20 depletion with a consequent loss of cilium-induced mislocalization of PDGFRα to the plasma membrane could impair negative feedback for pathway regulation. This is due to the destabilization and degradation of the E3 ubiquitin ligases [88]. Although PDGFRα is one of the most studied RTKs from a ciliary perspective, other RTKs have also been linked to the primary cilium in various ways, yet the significance and regulation mechanism of their ciliary localization requires further research. For example, phosphorylation and activation by brain-derived neurotrophic factor (BDNF) of its target receptor, TRKB, decreased upon loss of BBS4 expression in cultured cells, thereby linking ciliary TRKB signaling to neuronal phenotypes associated with BBS [89]. Retinal ganglion cell (RGC) primary cilium-concentrated IGF1R could be lost after the injury, reducing IGF1 potency. While regenerated RGCs relocated IGF1R to the primary cilium, which maintained their signaling competence and regenerative ability [90]. Knockdown of IFT88 suppressed ciliogenesis in mouse 3T3-L1 preadipocytes and human mesenchymal stem cells (MSCs) through suppression of the IGF1R-Akt-PPARγ signaling pathway [91, 92].Further research uncovered the mechanism that elongated cilia prevent caveolin-1- and/or GM3-positive lipid rafts from being assembled around the ciliary base where insulin receptor proteins accumulate, thereby inhibiting insulin-Akt signaling [93]. In summary, the ciliary RTKs in various cell types can control important cellular and physiological processes context-dependent via the same downstream signaling pathways (e.g., the MAPK, PI3K-AKT, and phospholipase C γ pathway).
In summary, present evidence show the essential role of primary cilium in regulating various signaling pathways that initiated and controlled EMT program, suggesting the potential link between primary cilium and EMT. In the next section, we will list the direct evidence that shows the link between them.
Direct evidence for ciliary signaling in EMT
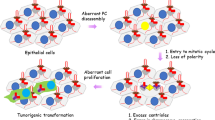
During the study of primary cilium, mutations in ciliary proteins result in loss of cell polarity [67]—an EMT feature and inhibit the progress of endothelial-to-mesenchymal transition (EndoMT), a similar program like EMT [94]. In addition, ciliary proteins can disrupt the expression of SIX1 [95], which is implicated in EMT [96]. Meanwhile loss of IFT88 lead to increased SNAIL by activating the WNT/β-catenin signaling pathway, which consequently decreased expression of E-cadherin and increased expression of vimentin in β-cells [20]. Similarly, the epicardial cells in mice expressing a loss-of-function mutant form of Wdpcp, an essential component for primary ciliogenesis, showed defective ciliogenesis and reduced expression of EMT and mesenchymal markers, causing increased distribution of SMO into subcellular sites, where chemotactic signaling can be transduced and increased chemotactic response, thereby leading to faster progression of subepicardial plexus in Wdpcp mutant although it showed normal SHH signaling transcriptional responses [22]. Most recently researchers studied the activation of EMT, HH signaling pathway and the presence of primary cilium in normal and cancer tissues by immunohistochemical and ultrastructural techniques. They found a correlation between EMT beginning from urothelial basal cells and primary cilia assembly and suggest a potential implication of this structure in tumoral migration and invasiveness (likely in a Hh-dependent way) [25].
Conversely, initial cilium growth is followed by complete deciliation during epithelial–myofibroblast transition (EMyT), a more extreme type of EMT that can occur in kidney epithelial cells [97]. Furthermore, TGFβ-induced shortening of primary cilium was found in Madin Darby Canine Kidney (MDCK) cells. The deficiency of primary cilium caused by knocking down the ciliary proteins Arl13b and IFT20 exacerbated TGFβ-induced EMT, suggesting positive feedback between TGFβ release, shortening or distortion of the primary cilium, and enhanced EMT/fibrosis in the kidney [19]. The relationship was further supported by Wilson MM et al., they showed that EMT-TFs promote ciliogenesis upon entry into intermediate EMT states. The resulting primary cilium promote ubiquitination and inactivation of a transcriptional repressor(GLIS2) to promote MaSC stemness and induce the proliferative and tumorigenic capacities of the mammary tumor initiating cells (MaTICs) of claudin-low breast cancers [16, 24].
Although there is no direct evidence that cilium can affect EMT through NOTCH signaling. RBPjk, the essential DNA binding partner of NOTCH receptors, was required to regulate mesothelial EMT and select Clara versus ciliated cell fate in lung development. In this research the authors described a distal-to-proximal transition zone in which ciliated cells induce Notch activation in their neighbors, inhibiting them from selecting the same fate and permitting development of Clara cells [98], providing the indirect link between cilium-based NOTCH pathway with EMT. In a recent study, Multiple binding motifs associated with SNAIL-dependent transcriptional regulation were identified in close proximity to or within the FGFR1 promoter, a key inducer of ciliogenesis in the embryonic tissues of lower organisms, by reanalyzing the data from an existing SNAIL chromatin immuno-precipitation sequencing (ChIP-seq) study [24]. In summary these evidence verify the relationship between primary cilium and EMT.
Conclusion
EMT is a remarkable mechanism that is essential for normal development, wound healing and carcinoma progression.
In the context of cancer, EMT and its reversed process, mesenchymal-to-epithelial transition (MET), confer additional malignant properties to cancer cells, including cancer stem cell activity [99] and more excellent resistance to chemotherapy and immunotherapy [100]. Previous research revealed that the presence or absence of the primary cilium could trigger or inhibit cancer progression depending on the cancer type and cancer-initiating mutations. The aberrant ciliogenesis could serve as a functional platform for a variety of cancer drug resistance mechanisms [101], suggesting that targeting primary cilium could be a promising treatment strategy. In addition, in the GBM and triple-negative breast cancer (TNBC) BAG3 knockout cells, enhanced ciliogenesis and reduced expression of SNAI1 and ZEB1 were correlated to decreased cell migration, suggesting that suppression of EMT and ciliogenesis as putative synergizing mechanisms of BAG3‐driven tumor aggressiveness in therapy‐resistant cancers [23].Understanding the ciliary signaling mechanisms that cooperate in the initiation and progression of EMT may lead to new therapeutic strategies to inhibit this cellular transformation in cancer. Nevertheless, limited studies were available to reveal the mechanism between primary cilium and EMT.
Primary cilium function as signal nexus to regulate signaling pathways that induce the initiation and progression of EMT, suggesting the potential link between primary cilium and EMT. Emerging evidence revealed the connection between EMT and primary cilium in multi organs and tissues, including epicardial tissue, kidney epithelial cells, pancreatic β-cells and the retinal pigment epithelium(RPE) [18,19,20,21,22] and in the context of cancer such as glioblastoma(GBM) and triple-negative breast cancer (TNBC) [23, 24], bladder cancer [25].But the relationship between primary cilium and EMT seem to be complicated and cell type, condition specific. For instance deficiency of primary cilium induces EMT in kidney epithelial cells [19, 97] and pancreatic β-cells [20], but inhibit EMT in the epicardial cells [22].These data prove that primary cilium function dual role upstream of EMT, but in the context of mammogenesis and claudin-low breast tumorigenesis, EMT programs induce primary ciliogenesis upon entry into intermediate transition states [24]. In addition, increased ciliogenesis caused by loss of BBS8 in RPE cells showed a EMT-like phenotype and the downstream EMT-TF SNAIL was only found in the nuclei of BBS8 mutant tissue [21].
In a word, emerging evidence has established the relationship between primary cilium and EMT under different conditions. Primary cilium interfere with EMT through signal transduction, but the mechanism is complex and puzzling and its details still need to be solved. The study of EMT axis of primary cilium has just begun and needs more effort.
Data availability
Not applicable.
References
Zhang Y, Weinberg RA (2018) Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med 12(4):361–373. https://doi.org/10.1007/s11684-018-0656-6
Nieto MA, Huang RY, Jackson RA, Thiery JP (2016) Emt: 2016. Cell 166(1):21–45. https://doi.org/10.1016/j.cell.2016.06.028
Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI et al (2016) Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res 365(3):495–506. https://doi.org/10.1007/s00441-016-2464-0
Huang Y, Hong W, Wei X (2022) The molecular mechanisms and therapeutic strategies of Emt in tumor progression and metastasis. J Hematol Oncol 15(1):129. https://doi.org/10.1186/s13045-022-01347-8
Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S et al (2018) Identification of the tumour transition states occurring during Emt. Nature 556(7702):463–468. https://doi.org/10.1038/s41586-018-0040-3
Pastushenko I, Blanpain C (2019) Emt transition states during tumor progression and metastasis. Trends Cell Biol 29(3):212–226. https://doi.org/10.1016/j.tcb.2018.12.001
Stemmler MP, Eccles RL, Brabletz S, Brabletz T (2019) Non-redundant functions of Emt transcription factors. Nat Cell Biol 21(1):102–112. https://doi.org/10.1038/s41556-018-0196-y
Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20(2):69–84. https://doi.org/10.1038/s41580-018-0080-4
Nachury MV, Mick DU (2019) Establishing and regulating the composition of cilia for signal transduction. Nat Rev Mol Cell Biol 20(7):389–405. https://doi.org/10.1038/s41580-019-0116-4
Carter SP, Blacque OE (2019) Membrane retrieval, recycling and release pathways that organise and sculpt the ciliary membrane. Curr Opin Cell Biol 59:133–139. https://doi.org/10.1016/j.ceb.2019.04.007
Wang J, Barr MM (2018) Cell-cell communication via ciliary extracellular vesicles: clues from model systems. Essays Biochem 62(2):205–213. https://doi.org/10.1042/EBC20170085
Wachten D, Mick DU (2021) Signal transduction in primary cilia—Analyzing and manipulating Gpcr and second messenger signaling. Pharmacol Ther 224:107836. https://doi.org/10.1016/j.pharmthera.2021.107836
Nishimura Y, Kasahara K, Shiromizu T, Watanabe M, Inagaki M (2019) Primary cilia as signaling hubs in health and disease. Adv Sci 6(1):1801138. https://doi.org/10.1002/advs.201801138
Picon-Galindo E, Latz E, Wachten D (2022) Primary cilia and their effects on immune cell functions and metabolism: a model. Trends Immunol 43(5):366–378. https://doi.org/10.1016/j.it.2022.03.001
Wang B, Liang Z, Liu P (2021) Functional aspects of primary cilium in signaling, assembly and microenvironment in cancer. J Cell Physiol 236(5):3207–3219. https://doi.org/10.1002/jcp.30117
Guen VJ, Chavarria TE, Kroger C, Ye X, Weinberg RA, Lees JA (2017) Emt programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and hedgehog signaling. Proc Natl Acad Sci U S A 114(49):E10532–E10539. https://doi.org/10.1073/pnas.1711534114
Eguether T, Hahne M (2018) Mixed signals from the cell’s antennae: primary cilia in cancer. EMBO Rep. https://doi.org/10.15252/embr.201846589
Blom JN, Feng Q (2018) Cardiac repair by epicardial Emt: current targets and a potential role for the primary cilium. Pharmacol Ther 186:114–129. https://doi.org/10.1016/j.pharmthera.2018.01.002
Han SJ, Jung JK, Im SS, Lee SR, Jang BC, Park KM et al (2018) Deficiency of primary cilia in kidney epithelial cells induces epithelial to mesenchymal transition. Biochem Biophys Res Commun 496(2):450–454. https://doi.org/10.1016/j.bbrc.2018.01.079
Volta F, Scerbo MJ, Seelig A, Wagner R, O’Brien N, Gerst F et al (2019) Glucose homeostasis is regulated by pancreatic beta-cell cilia via endosomal Epha-processing. Nat Commun 10(1):5686. https://doi.org/10.1038/s41467-019-12953-5
Schneider S, De Cegli R, Nagarajan J, Kretschmer V, Matthiessen PA, Intartaglia D et al (2021) Loss of ciliary gene Bbs8 results in physiological defects in the retinal pigment epithelium. Front Cell Dev Biol 9:607121. https://doi.org/10.3389/fcell.2021.607121
Liu X, Wang Y, Liu F, Zhang M, Song H, Zhou B et al (2018) Wdpcp Promotes epicardial Emt and epicardium-derived cell migration to facilitate coronary artery remodeling. Sci Signal. https://doi.org/10.1126/scisignal.aah5770
Linder B, Klein C, Hoffmann ME, Bonn F, Dikic I, Kogel D (2022) Bag3 is a negative regulator of ciliogenesis in glioblastoma and triple-negative breast cancer cells. J Cell Biochem 123(1):77–90. https://doi.org/10.1002/jcb.30073
Wilson MM, Callens C, Le Gallo M, Mironov S, Ding Q, Salamagnon A et al (2021) An Emt-primary cilium-Glis2 signaling axis regulates mammogenesis and claudin-low breast tumorigenesis. Sci Ad 7(44):063. https://doi.org/10.1126/sciadv.abf6063
Iruzubieta P, Castiella T, Monleon E, Berga C, Munoz G, Junquera C (2021) Primary cilia presence and implications in bladder cancer progression and invasiveness. Histochem Cell Biol 155(5):547–560. https://doi.org/10.1007/s00418-021-01965-2
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196. https://doi.org/10.1038/nrm3758
Fabbri L, Bost F, Mazure NM (2019) Primary cilium in cancer hallmarks. Int J Mol Sci. https://doi.org/10.3390/ijms20061336
Massagué J (2012) Tgfβ signalling in context. Nat Rev Mol Cell Biol 13(10):616–630. https://doi.org/10.1038/nrm3434
Clement Christian A, Ajbro Katrine D, Koefoed K, Vestergaard Maj L, Veland Iben R, de Jesus H, Maria Perestrello R et al (2013) Tgf-Β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep 3(6):1806–1814. https://doi.org/10.1016/j.celrep.2013.05.020
Koefoed K, Skat-Rordam J, Andersen P, Warzecha CB, Pye M, Andersen TA et al (2018) The E3 ubiquitin ligase Smurf1 regulates cell-fate specification and outflow tract septation during mammalian heart development. Sci Rep 8(1):9542. https://doi.org/10.1038/s41598-018-27854-8
Labour MN, Riffault M, Christensen ST, Hoey DA (2016) Tgfbeta1—Induced recruitment of human bone mesenchymal stem cells is mediated by the primary cilium in a Smad3-dependent manner. Sci Rep 6:35542. https://doi.org/10.1038/srep35542
Gencer S, Oleinik N, Kim J, Panneer Selvam S, De Palma R, Dany M et al (2017) Tgf-Beta Receptor I/Ii trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci Signal. https://doi.org/10.1126/scisignal.aam7464
Villalobos E, Criollo A, Schiattarella GG, Altamirano F, French KM, May HI et al (2019) Fibroblast primary cilia are required for cardiac fibrosis. Circulation 139(20):2342–2357. https://doi.org/10.1161/CIRCULATIONAHA.117.028752
Liu M, Alharbi M, Graves D, Yang S (2020) Ift80 is required for fracture healing through controlling the regulation of Tgf-beta signaling in chondrocyte differentiation and function. J Bone Miner Res 35(3):571–582. https://doi.org/10.1002/jbmr.3902
Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L et al (2011) Primary cilia membrane assembly is initiated by Rab11 and transport protein particle Ii (Trappii) complex-dependent trafficking of rabin8 to the centrosome. Proc Natl Acad Sci U S A 108(7):2759–2764. https://doi.org/10.1073/pnas.1018823108
Monnich M, Borgeskov L, Breslin L, Jakobsen L, Rogowski M, Doganli C et al (2018) Cep128 localizes to the subdistal appendages of the mother centriole and regulates Tgf-Beta/Bmp signaling at the primary cilium. Cell Rep 22(10):2584–2592. https://doi.org/10.1016/j.celrep.2018.02.043
Zhang X, Wang L, Ma Y, Wang Y, Liu H, Liu M et al (2022) Cep128 is involved in spermatogenesis in humans and mice. Nat Commun 13(1):1395. https://doi.org/10.1038/s41467-022-29109-7
Ehnert S, Sreekumar V, Aspera-Werz RH, Sajadian SO, Wintermeyer E, Sandmann GH et al (2017) Tgf-beta(1) impairs mechanosensation of human osteoblasts via Hdac6-mediated shortening and distortion of primary cilia. J Mol Med 95(6):653–663. https://doi.org/10.1007/s00109-017-1526-4
Kawasaki M, Ezura Y, Hayata T, Notomi T, Izu Y, Noda M (2015) Tgf-beta suppresses ift88 expression in chondrocytic Atdc5 cells. J Cell Physiol 230(11):2788–2795. https://doi.org/10.1002/jcp.25005
Ho EK, Stearns T (2021) Hedgehog signaling and the primary cilium: implications for spatial and temporal constraints on signaling. Development. https://doi.org/10.1242/dev.195552
Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426:83–87. https://doi.org/10.1038/nature02061
Larkins CE, Aviles GD, East MP, Kahn RA, Caspary T (2011) Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell 22(23):4694–4703. https://doi.org/10.1091/mbc.E10-12-0994
Bay SN, Long AB, Caspary T (2018) Disruption of the ciliary Gtpase Arl13b suppresses sonic hedgehog overactivation and inhibits medulloblastoma formation. Proc Natl Acad Sci U S A 115(7):1570–1575. https://doi.org/10.1073/pnas.1706977115
Truong ME, Bilekova S, Choksi SP, Li W, Bugaj LJ, Xu K et al (2021) Vertebrate cells differentially interpret ciliary and extraciliary camp. Cell 184(11):2911–26 e18. https://doi.org/10.1016/j.cell.2021.04.002
Ho EK, Tsai AE, Stearns T (2020) Transient primary cilia mediate robust hedgehog pathway-dependent cell cycle control. Curr Biol 30(14):2829–35 e5. https://doi.org/10.1016/j.cub.2020.05.004
Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317(5836):372–376. https://doi.org/10.1126/science.1139740
Goetz SC, Ocbina PJ, Anderson KV (2009) The primary cilium as a hedgehog signal transduction machine. Methods Cell Biol 94:199–222. https://doi.org/10.1016/S0091-679X(08)94010-3
Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ et al (2013) The ciliary G-protein-coupled receptor Gpr161 negatively regulates the sonic hedgehog pathway via camp signaling. Cell 152(1–2):210–223. https://doi.org/10.1016/j.cell.2012.12.026
Nager AR, Goldstein JS, Herranz-Perez V, Portran D, Ye F, Garcia-Verdugo JM et al (2017) An actin network dispatches ciliary Gpcrs into extracellular vesicles to modulate signaling. Cell 168(1–2):252-263 e14. https://doi.org/10.1016/j.cell.2016.11.036
Pal K, Hwang SH, Somatilaka B, Badgandi H, Jackson PK, DeFea K et al (2016) Smoothened determines beta-arrestin-mediated removal of the G protein-coupled receptor Gpr161 from the primary cilium. J Cell Biol 212(7):861–875. https://doi.org/10.1083/jcb.201506132
Jaszai J, Thamm K, Karbanova J, Janich P, Fargeas CA, Huttner WB et al (2020) Prominins control ciliary length throughout the animal kingdom: new lessons from human prominin-1 and zebrafish prominin-3. J Biol Chem 295(18):6007–6022. https://doi.org/10.1074/jbc.RA119.011253
Singer D, Thamm K, Zhuang H, Karbanova J, Gao Y, Walker JV et al (2019) Prominin-1 controls stem cell activation by orchestrating ciliary dynamics. EMBO J. https://doi.org/10.15252/embj.201899845
Yuan X, Cao J, He X, Serra R, Qu J, Cao X et al (2016) Ciliary Ift80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat Commun 7:11024. https://doi.org/10.1038/ncomms11024
Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr et al (2009) Primary cilia can both mediate and suppress hedgehog pathway-dependent tumorigenesis. Nat Med 15(9):1055–1061. https://doi.org/10.1038/nm.2011
Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15(9):1062–1065. https://doi.org/10.1038/nm.2020
Jacob LS, Wu X, Dodge ME, Fan CW, Kulak O, Chen B et al (2011) Genome-wide Rnai screen reveals disease-associated genes that are common to hedgehog and Wnt signaling. Sci Signal 4(157):ra4. https://doi.org/10.1126/scisignal.2001225
Parsons MJ, Tammela T, Dow LE (2021) Wnt as a driver and dependency in cancer. Cancer Discov 11(10):2413–2429. https://doi.org/10.1158/2159-8290.CD-21-0190
Perkins RS, Suthon S, Miranda-Carboni GA, Krum SA (2022) Wnt5b in cellular signaling pathways. Semin Cell Dev Biol 125:11–16. https://doi.org/10.1016/j.semcdb.2021.09.019
Zhang B, Zhang T, Wang G, Wang G, Chi W, Jiang Q et al (2015) Gsk3beta-Dzip1-Rab8 cascade regulates ciliogenesis after mitosis. PLoS Biol 13(4):e102129. https://doi.org/10.1371/journal.pbio.1002129
Marion V, Stoetzel C, Schlicht D, Messaddeq N, Koch M, Flori E et al (2009) Transient ciliogenesis involving bardet-biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci U S A 106(6):1820–1825. https://doi.org/10.1073/pnas.0812518106
Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C et al (2005) Inversin, the gene product mutated in nephronophthisis type Ii, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37(5):537–543. https://doi.org/10.1038/ng1552
Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH et al (2008) Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 10(1):70–76. https://doi.org/10.1038/ncb1670
Lancaster MA, Schroth J, Gleeson JG (2011) Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol 13(6):700–707. https://doi.org/10.1038/ncb2259
Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M et al (2007) Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 39(11):1350–1360. https://doi.org/10.1038/ng.2007.12
Patnaik SR, Kretschmer V, Brucker L, Schneider S, Volz AK, Oancea-Castillo LDR et al (2019) Bardet-Biedl syndrome proteins regulate cilia disassembly during tissue maturation. Cell Mol Life Sci 76(4):757–775. https://doi.org/10.1007/s00018-018-2966-x
Li Y, Zhang X, Polakiewicz RD, Yao T-P, Comb MJ (2008) Hdac6 is required for epidermal growth factor-induced β-catenin nuclear localization. J Biol Chem 283(19):12686–12690. https://doi.org/10.1074/jbc.C700185200
Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ et al (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37(10):1135–1140. https://doi.org/10.1038/ng1644
May-Simera HL, Kai M, Hernandez V, Osborn DP, Tada M, Beales PL (2010) Bbs8, together with the planar cell polarity protein Vangl 2, is required to establish left-right asymmetry in zebrafish. Dev Biol 345(2):215–225. https://doi.org/10.1016/j.ydbio.2010.07.013
May-Simera HL, Petralia RS, Montcouquiol M, Wang YX, Szarama KB, Liu Y et al (2015) Ciliary proteins Bbs8 and Ift20 Promote planar cell polarity in the cochlea. Development 142(3):555–566. https://doi.org/10.1242/dev.113696
May-Simera HL, Wan Q, Jha BS, Hartford J, Khristov V, Dejene R et al (2018) Primary cilium-mediated retinal pigment epithelium maturation is disrupted in ciliopathy patient cells. Cell Rep 22(1):189–205. https://doi.org/10.1016/j.celrep.2017.12.038
Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL et al (2009) Convergent extension movements and ciliary function are mediated by Ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet 18(2):289–303. https://doi.org/10.1093/hmg/ddn356
Volz AK, Frei A, Kretschmer V, de Jesus Domingues AM, Ketting RF, Ueffing M et al (2021) Bardet-Biedl syndrome proteins modulate the release of bioactive extracellular vesicles. Nat Commun 12(1):5671. https://doi.org/10.1038/s41467-021-25929-1
Nowell CS, Radtke F (2017) Notch as a tumour suppressor. Nat Rev Cancer 17(3):145–159. https://doi.org/10.1038/nrc.2016.145
Aster JC, Pear WS, Blacklow SC (2017) The varied roles of notch in cancer. Annu Rev Pathol 12:245–275. https://doi.org/10.1146/annurev-pathol-052016-100127
Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E (2011) A Role for the primary cilium in notch signaling and epidermal differentiation during skin development. Cell 145(7):1129–1141. https://doi.org/10.1016/j.cell.2011.05.030
Ezratty EJ, Pasolli HA, Fuchs E (2016) A Presenilin-2-Arf4 trafficking axis modulates notch signaling during epidermal differentiation. J Cell Biol 214(1):89–101. https://doi.org/10.1083/jcb.201508082
Grisanti L, Revenkova E, Gordon RE, Iomini C (2016) Primary cilia maintain corneal epithelial homeostasis by regulation of the notch signaling pathway. Development 143(12):2160–2171. https://doi.org/10.1242/dev.132704
Li X, Lu Q, Peng Y, Geng F, Shao X, Zhou H et al (2020) Primary cilia mediate Klf2-dependant notch activation in regenerating heart. Protein Cell 11(6):433–445. https://doi.org/10.1007/s13238-020-00695-w
Liu Z, Tu H, Kang Y, Xue Y, Ma D, Zhao C et al (2019) Primary cilia regulate hematopoietic stem and progenitor cell specification through notch signaling in zebrafish. Nat Commun 10(1):1839. https://doi.org/10.1038/s41467-019-09403-7
Leitch CC, Lodh S, Prieto-Echague V, Badano JL, Zaghloul NA (2014) Basal body proteins regulate notch signaling through endosomal trafficking. J Cell Sci 127(Pt 11):2407–2419. https://doi.org/10.1242/jcs.130344
Du Z, Lovly CM (2018) Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 17(1):58. https://doi.org/10.1186/s12943-018-0782-4
Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST (2019) Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol 15(4):199–219. https://doi.org/10.1038/s41581-019-0116-9
Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P et al (2005) PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15(20):1861–1866. https://doi.org/10.1016/j.cub.2005.09.012
Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR et al (2010) Directional cell migration and chemotaxis in wound healing response to Pdgf-Aa are coordinated by the primary cilium in fibroblasts. Cell Physiol Biochem 25(2–3):279–292. https://doi.org/10.1159/000276562
Goranci-Buzhala G, Mariappan A, Ricci-Vitiani L, Josipovic N, Pacioni S, Gottardo M et al (2021) Cilium induction triggers differentiation of glioma stem cells. Cell Rep 36(10):109656. https://doi.org/10.1016/j.celrep.2021.109656
Suizu F, Hirata N, Kimura K, Edamura T, Tanaka T, Ishigaki S et al (2016) Phosphorylation-dependent Akt-inversin interaction at the basal body of primary cilia. EMBO J 35(12):1346–1363. https://doi.org/10.15252/embj.201593003
Guo DF, Rahmouni K (2019) The Bardet-Biedl syndrome protein complex regulates cell migration and tissue repair through a Cullin-3/Rhoa pathway. Am J Physiol Cell Physiol 317(3):C457–C465. https://doi.org/10.1152/ajpcell.00498.2018
Schmid FM, Schou KB, Vilhelm MJ, Holm MS, Breslin L, Farinelli P et al (2018) Ift20 modulates ciliary Pdgfrα Signaling by regulating the stability of Cbl E3 ubiquitin ligases. J Cell Biol 217(1):151–161. https://doi.org/10.1083/jcb.201611050
Leitch CC, Zaghloul NA (2014) Bbs4 is necessary for ciliary localization of Trkb receptor and activation by Bdnf. PLoS ONE 9(5):e98687. https://doi.org/10.1371/journal.pone.0098687
Zhang Y, Williams PR, Jacobi A, Wang C, Goel A, Hirano AA et al (2019) Elevating growth factor responsiveness and axon regeneration by modulating presynaptic inputs. Neuron 103(1):39-51 e5. https://doi.org/10.1016/j.neuron.2019.04.033
Zhu D, Shi S, Wang H, Liao K (2009) Growth arrest induces primary-cilium formation and sensitizes Igf-1-receptor signaling during differentiation induction of 3t3-L1 preadipocytes. J Cell Sci 122(Pt 15):2760–2768. https://doi.org/10.1242/jcs.046276
Dalbay MT, Thorpe SD, Connelly JT, Chapple JP, Knight MM (2015) Adipogenic differentiation of hMSCs is mediated by recruitment of Igf-1r onto the primary cilium associated with cilia elongation. Stem Cells 33(6):1952–1961. https://doi.org/10.1002/stem.1975
Yamakawa D, Katoh D, Kasahara K, Shiromizu T, Matsuyama M, Matsuda C et al (2021) Primary cilia-dependent lipid Raft/Caveolin dynamics regulate adipogenesis. Cell Rep 34(10):108817. https://doi.org/10.1016/j.celrep.2021.108817
Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P et al (2011) Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res 108(9):1093–1101. https://doi.org/10.1161/CIRCRESAHA.110.231860
Gascue C, Tan PL, Cardenas-Rodriguez M, Libisch G, Fernandez-Calero T, Liu YP et al (2012) Direct role of bardet-biedl syndrome proteins in transcriptional regulation. J Cell Sci 125(Pt 2):362–375. https://doi.org/10.1242/jcs.089375
Micalizzi DS, Christensen KL, Jedlicka P, Coletta RD, Baron AE, Harrell JC et al (2009) The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing Tgf-beta signaling. J Clin Investig 119(9):2678–2690. https://doi.org/10.1172/JCI37815
Rozycki M, Lodyga M, Lam J, Miranda MZ, Fatyol K, Speight P et al (2014) The fate of the primary cilium during myofibroblast transition. Mol Biol Cell 25(5):643–657. https://doi.org/10.1091/mbc.E13-07-0429
Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R (2010) Canonical notch signaling in the develo** lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci 123(Pt 2):213–224. https://doi.org/10.1242/jcs.058669
Babaei G, Aziz SG, Jaghi NZZ (2021) Emt, cancer stem cells and autophagy; the three main axes of metastasis. Biomed Pharmacother 133:110909. https://doi.org/10.1016/j.biopha.2020.110909
De Las RJ, Brozovic A, Izraely S, Casas-Pais A, Witz IP, Figueroa A (2021) Cancer drug resistance induced by Emt: novel therapeutic strategies. Arch Toxicol 95(7):2279–2297. https://doi.org/10.1007/s00204-021-03063-7
Jenks AD, Vyse S, Wong JP, Kostaras E, Keller D, Burgoyne T et al (2018) Primary cilia mediate diverse kinase inhibitor resistance mechanisms in cancer. Cell Rep 23(10):3042–3055. https://doi.org/10.1016/j.celrep.2018.05.016
Acknowledgements
Not applicable
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Corresponding author MD contributed to the conception and design of the study, first author BHZ wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhong, BH., Dong, M. The implication of ciliary signaling pathways for epithelial–mesenchymal transition. Mol Cell Biochem (2023). https://doi.org/10.1007/s11010-023-04817-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11010-023-04817-w