
Abstract
Heat shock protein 70 kDa (HSP70) is a major protein family in the cell protections against stress-induced denaturation and aggregation and in the folding of nascent proteins. It is a highly conserved protein that can be found in most organisms and is strongly connected to several intracellular pathways such as protein folding and refolding, protein degradation and regulation, and protection against intense stress. Cellular delivery of HSP70 would be of high impact for clarification of its role in these cellular processes.
PepFect14 is a cell-penetrating peptide known to be able to mediate the transfection of various oligonucleotides to multiple cell lines with a higher efficacy than most commercially available transfection agents and without inducing significant toxic effects.
In this study we demonstrated that PepFect14 was able to form a complex with HSP70 and to deliver it inside cells in the same fashion with oligonucleotide delivery. The delivered HSP70 showed an effect in the cell regulation indicating that the protein was biologically available in the cytoplasm and the interactions with PepFect14 did not impeach its active sites once the plasma barrier crossed.
This study reports the first successful delivery of HSP70 to our knowledge and the first protein transfection mediated by PepFect14. It opens new fields of research for both PepFect14 as a delivery agent and HSP70 as a therapeutic agent; with potential in peptide aggregation caused diseases such as Parkinson’s and Alzheimer’s diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heat shock protein 70 kDa (HSP70) is a highly conserved protein family expressed by most organisms and found in both prokaryotes and eukaryotes. All the members of the HSP70 protein family have a molecular weight that ranges around 70 kDa and all share the same chaperone function (Radons 2016). They bind to short hydrophobic polypeptide sequences in their substrate binding domain (SBD) with an affinity that is allosterically modulated by the N-terminal adenine nucleotide binding domain (NBD) (Feige and Polla 1994). When adenosine triphosphate is bound to the NBD the affinity for the substrate in the SBD is low but when adenosine diphosphate is bound, the affinity increases (Clerico et al. 2015). This process actually triggers the closing of the C-terminal α-helical domain. This domain acts as a lid that retains the substrate bound to the rigid structure of the SBD (Strub et al. 2003). HSP70 has two major roles in the cells; first, it binds to peptidic parts of nascent proteins and keeps them from premature folding (Bukau et al. 2000; Hartl and Hayer-Hartl 2002); second, HSP70 has a protective feature in response to intense cellular stress such as environmental, physiological or pathological stress (Pinto et al. 1991; Qu et al. 2015). HSP70 is of major importance in the regulation and protection of intracellular proteins. Indeed, HSP70, besides being closely involved in the folding of newly produced proteins as a chaperone protein, is also involved in many other intracellular regulative pathways. When a substrate binds to HSP70, several pathways can be triggered (Kim et al. 2018). The substrate can be refolded, either directly by the action of HSP70 or through the recruitment of co-chaperones (Sharma and Masison 2009), but can also be translocated to autophagosome or proteasome for intracellular recycling (Witkin et al. 2017). HSP70 can also trigger regulations of transcription factors that lead to an indirect regulation of the proteome (Pratt and Toft 2003). Under an intense stress, HSP70 is able to bind to already formed proteins and to protect them against denaturation and aggregation (Rokutan 2000). All these possible pathways triggered by HSP70 give this protein a major role in maintaining cell functions. HSP70 can also play an important role as a therapeutic agent in neurodege- nerative diseases caused by peptide aggregations such as α-synuclein in Parkinson’s disease (Rochet et al. 2012) or amyloid-β and Tau in Alzheimer’s disease (Campanella et al. 2018).
Modulating intracellular levels of HSP70 has the potential to be an efficient therapeutic action (Kim et al. 2018) and more particularly an acute increase of its concentration could give a temporary protection against protein aggregation and misfolding. There are two methods to increase the level of a specific protein in cells. Either transfecting an oligonucleotide or a plasmid DNA coding for this protein (Kim and Eberwine 2010) or delivering directly the wanted protein inside the cell (Pisal et al. 2010). The transfection of oligonucleotides and plasmid DNA can be either permanent if the sequence is integrated in the genome or transient if not. In either case, the expression of the protein relies on the cell machinery and is dependent on transcription factor. If a protein is delivered instead, it is directly available for a biological effect the delivery will then be transient as the cells will not express the protein after its natural degradation. Delivering a protein is an alternative to oligonucleotide and plasmid DNA delivery and presents advantages for acute and non-permanent treatment. It also allows a more straight-forward treatment strategy as it does not rely on the cell transcription machinery (Lee et al. 2019).
Cell-penetrating peptides (CPPs) are a class of peptides able to translocate through biological barriers and to transport bioactive macromolecules directly inside cells (Bechara and Sagan 2013). PepFect14 is an amphipathic CPP composed by a 21 amino acids sequence and a stearic acid tail bound to its N-terminus (Ezzat et al. 2011) with sequence stearoyl-AGYLLGKLLOOLAAAALOOLL. It is able to form non covalent complexes with its cargoes via electrostatic interactions (Lehto et al. 2017). It has been shown for several years already that PepFect14 formed complexes with diverse modified or non-modified oligonucleotides – siRNA (Ervin et al. 2019), plasmid DNA (Veiman et al. 2013) and antisense oligonucleotides (Ezzat et al. 2011) - and mediated their transfection inside various types of cells. The complexes, when placed in cell culture media, formed nanoparticle with a hydrodynamic diameter in the range of 102 nm (Lehto et al. 2017). It was shown in a previous study from our group that the formation of nanoparticles was a key factor in the ability of PepFect14 to mediates transfections (Lehto et al. 2016). In another study where we studied the gene regulations induced by the translocation of PepFect14 in HeLa cells, we demonstrated that HSPA1B, a gene that codes for a HSP70 protein, was up-regulated as a reaction to the uptake of PepFect14. In the same study, we showed that a binding opportunity between PepFect14 and HSP70 existed in the polypeptide binding site (Dowaidar et al. 2017).
In the present study, we assessed the ability of PepFect14 to form a complex with HSP70. We also characterized the particles that these complexes formed in cell culture media before monitoring the uptake of a fluorescent-labelled HSP70 in cells and finally we detected a biological effect induced by the transfected HSP70.
Materials and Methods
Peptide Synthesis
PepFect14’s amino acid chain was synthesized using a Biotage Alstra + microwave assisted synthe- sizer (Biotage, Sweden) on a 0.1 mmol scale following fluorenylmethyloxycarbonyl (Fmoc) solid phase peptide synthesis chemistry. The first amino acid was bound to a ChemMatrix Rink Amide resin (0.50 mmol/g) as a solid support in order to get a C-terminal amide group. The resin was swollen in N,N-dimethylformamide (DMF) (VWR, Radnor, Pennsylvania, USA) for 5 min at 70°C with oscillation mixer on. Fmoc-protected amino acids were dissolved in DMF containing 2-cyano-2-(hydroxyimi- no)acetate (OxymaPure, Novabio- chem, Merck Millipore, Burlington, Massachusetts, USA) and N,N’-Diisopropylcarbodiimide (DIC) (Iris Biotech GmbH, Germany) as coupling reagents and added to the resin for 5 min at 75 °C. The Fmoc protective group was removed with 20% piperidine (Iris Biotech GmbH, Germany) in DMF for 2 min at 45 °C followed by a 12 min reaction at room temperature before addition of the next amino acid. The final cleavage was performed with 95% trifluoroacetic acid (TFA) (Iris Biotech GmbH, Germany), 2.5% triisopropylsilane (TIS) (Merck KGaA, Darmstadt, Germany) and 2.5% water for 4 h at room temperature before precipitation in cold diethyl ether (VWR, Radnor, Pennsylvania, USA). The crude peptide was dissolved in 80% acetonitrile (VWR, Radnor, Pennsylvania, USA) and 20% water before purification by reverse-phase high performance liquid chromatography (RP-HPLC) using a Biobasic C8 column (Thermo Scientific, Sweden) with a gradient of acetonitrile in water (0.1% TFA). The purified peptide was freeze-dried and the molecular mass was verified by UHPLC-MS (Agilent 1260 Infinity, Agilent Technologies, Santa Clara, California, USA).
Protein Expression and Purification and Labelling
Plasmid pCA1033 was constructed by previously described approach (Holmberg et al., 2014). Briefly, the Hsp70 gene (HSPA1A) was PCR amplified from pcDNA/FRT/TO-V5-HSPA1A (Hageman and Kam**a, 2009). The PCR amplified product was used to transform yeast together with the restricted vector pSUMO-YHRC to yield pCA1033 using yeast homologous recombination. The plasmid was rescued to E. coli and was verified by DNA sequencing.
Hsp70 was expressed in E. coli strain BL21-SI/pCodonPlus from plasmid pCA1033 as a 6xHis-SUMO fusion as described previously (Andréasson et al. 2008). Briefly, an overnight culture of the transformed cells was diluted 100-fold in 1 L 2xYTON supplemented with 50 mg/L kanamycin, 25 mg/L chloramphe- nicol and 2 mM MgSO4 and expression of the protein was induced by the addition of 1 mL 0.5 M iso-propyl b-D-thiogalactopyrano- side (IPTG) and 40 mL 5 M NaCl at OD600 0.6. After 6 h expression at 30 °C cells were harvested by centrifugation and the pellet was resuspended in 10 mL of lysis buffer LWB150 (40 mM HEPES pH 7.4, 150 mM KCl, 5 mM MgCl2, 5% glycerol) supplemented with 1 mM PMSF and DNase I and complete (TM) EDTA-free protease inhibit and lysed by two passages in an EmulsiFlex-C3 high-pressure homogenizer. The cleared supernatant (27,000 g for 30 min) was incubated with Protino Ni-IDA silica matrix (Machery-Nagel GmbH and Co. KG, Düren, Germany) for 1 h at 4 °C with end over rotation. The matrix was transferred to a gravity-flow column and after extensive washing with LWB150 the bound protein was eluted using LWB150 supplemented with 250 mM imidazole pH 7.4. The 6xHis-SUMO was cleaved with Ulp1-6xHis protease during overnight dialysis against LWB150 at 4 °C. The dialyzed solution was passed over Ni-IDA matrix to remove 6xHis-SUMO and Ulp1-6xHis. For microscopy studies, the part of the purified proteins was labelled with Alexa 568 fluorophore using Alexa Fluor™ 568 Protein Labeling Kit (Invitrogen, Sweden) according to manufacturer protocol.
Tryptophan Fluorescence Extinction
The formation of a complex between PepFect14 and HSP70 was investigated by tryptophan fluorescence extinction. In a black opaque 96-well plate, HSP70 (50 nM) was incubated in HEPES buffer (1mM) in presence of various concentration of PepFect14 (from 1 nM to 1 µM) for 30 min at 37 °C. The plate was then read on a fluorescence reader (Flex Station II, Molecular Device; ex: 280 nm, em: 360 nm). The measurement was made in triplicates for each PepFect14 concentration. The fluorescence intensities were then transformed and normalized to the intensity of HSP70 alone (Eq. 1, where I represent the intensity of tryptophan fluorescence) to obtain the percentage of fluorescence extinction.
The data were analysed using GraphPad Prism 8 and were fitted with a hyperbolic regression to obtain an apparent Kd value.
Dynamic Light Scattering
The particle formation ability of our complexes PF14:HSP70 was assessed using a Zetasizer Nano ZS (Malvern Instruments, United Kingdom). The hydrodynamic diameter of PepFect14 (2 µM), HSP70 (200 nM) and PF14:HSP70 (2µM:200nM) were measured at 37 °C in DMEM supplemented with 10% FBS, to fit to the cell treatment conditions. The compound were prepared in 60 µL milliQ water 10 time more concentrated and then diluted to 600 µL in DMEM + 10% FBS before being transferred into DTS1070 cuvettes. PepFect14 and HSP70 complexes were let to incubate for 30 min at room temperature before dilution and reading.
Cell Culture
Bormirski hamster melanoma (BHM) cells stably expressing firefly luciferase (BHM pLuc cells) were grown in Dubelco’s Modifed Eagle Medium (DMEM) at 37 °C in humidified incubator with 5% CO2. The culture media was supplemented with 10% Fetal Bovine Serum (FBS), 200 µg/ml streptomycin and 200U/ml penicillin (Invitrogen, Sweden) and Plasmocin™ Prophylactic 5 mg/L (Invivogen, France).
Epi-Fluorescence Microscopy
Optical microscopy 96-well plates were seeded with 7,000 BHM pLuc expressing cells per well in 100 µL DMEM supplemented with 10% FBS and incubated at 37 °C for one day. The complex PF14:HSP70-Alexa 568 (2 µM:200 nM) were formed in milliQ water for 30 min at room temperature before addition to the cells for 1 or 3 h. A control experiment was prepared with HSP70-Alexa 568 alone (200 nM) incubated for 1 and 3 h. The wells were emptied and wash thrice with 200 µL Gibco Opti-MEM before a final addition of 100µL of Gibco FluoroBrite. The plate was then imaged using an epi-fluorescence microscope. The imaging was performed using a Leica DM/IRBE 2 epi-fluorescence microscope with a 63 × 1.4 NA oil immersion objective for fluorescence imaging, and the images were recorded by a Hamamatsu Orca-ER CCD camera. The system was controlled by the open source software Micro-Manager. Fluorescent images were collected using N3 filter cube (Chroma Technology Corporation, VT, USA). The surface plots were obtained using Image J software.
Luciferase Recovery
In opaque 96-well plates with clear bottom, 7,000 cells were seeded in DMEM with 10% FBS and incubated overnight at 37 °C. Lipofectamine 2000 (Thermofisher Scientific, Sweden) was incubated with siRNA against HSPA1A/B according to manufacturer protocols and added to the cells. The plates were further incubated for one day. On the third day, PepFect14 (20 µM) and HSP70 (2 µM) were mixed in milliQ water and left to form complexes for 30 min at room temperature. In each wells, except controls, PF14:HSP70 was added to fresh medium with dilution factor 10 and the cells were incubated for 3 more hours at 37 °C. The plates were then emptied and frozen at -80 °C overnight before thawing would complete the cell lysis. A luciferase assay was performed as described in Helmfors et al. (Helmfors et al. 2015). Briefly, a mixture of D-luciferin, adenosine triphosphate, co-enzyme A and dithiothreitol was prepared in buffer containing MgSO4, MgCO3, tricine and ethylenediaminetetraacetic acid. The luciferase assay mix was added to each well (100 µL per well) and the plates were shaken for 1 min before the luminescence was read in a luminometer.
Cell Proliferation Assay
To assess the possible toxic effects of our treatments, a WST-1 assay was performed during the same treatment as the ones used to measure the luciferase activity recovery. 10 µL of WST-1 reagent were added to the medium in each well and incubated for 2 h at 37 °C. The absorbance of each well was measured at 440 nm over 690 nm. The cell medium with WST-1 was then removed and the cells were lysed for the luciferase assay to be performed.
Results
HSP70 Forms a Complex with PepFect14
The binding of PepFect14 to HSP70 was assessed using a tryptophan fluorescence extinction assay. Tryptophan is fluorescent amino acids that absorbs and emits in the ultraviolet range of energy. The intensity of its fluorescence is highly correlated to its direct environment (Ghisaidoobe and Chung 2014). Thus, measuring the extinction of tryptophan fluorescence of a protein in presence of another compound gives information on the direct neighbouring of the residues (Vivian and Callis 2001). Indeed when a molecule is complexed to a protein, it results in a refolding of the structure that, in turns, changes the environment of the tryptophan residues and the intensity of the protein intrinsic fluorescence. This method allows the identification of protein binding using intrinsic protein properties. It avoids using additional fluorescent probes that would most likely already induce changes in the protein structure and thus is a straightforward and efficient technique to observe a non-covalent binding to a protein (Sindrewicz et al. 2019). We prepared HSP70 samples (50 nM) mixed with a dilution series of PepFect14 (from 1 µM to 1 nM) and recorded the changes in tryptophan fluorescence intensity. The data were transformed using Eq. 1 to obtain the percentage of extinction and the plot displayed in Fig. 1 A clearly showed a PepFect14 dose dependent extinction of HSP70 intrinsic tryptophan fluorescence. This extinction indicated a non-covalent interaction between our peptide and the protein. The curve was fitted with a hyperbolic regression in order to calculate the strength of the binding. The apparent Kd from this binding curve was calculated to be 34,98 nM and a maximum binding was observed at molar ratio 3. The complexes formed by PepFect14 and HSP70 will be noted PF14:HSP70 in the rest of this study.

A. Tryptophan fluorescence extinction. In HEPES buffer, HSP70 (50 nM) was put in presence of various concentration of PepFect14 (from 1 µM to 1 nM). Fluorescence intensities were measures with an excitation peak at 280 nm and an emission peak at 360 nm. The data were normalized to the intensity of HSP70 alone and to the buffer before being transformed to a percentage of extinction. Each data point shows the mean and SEM of three technical replicates. B. hydrodynamic diameter measurement by dynamic light scattering of the partiles present in DMEM + 10% FBS alone or supplemented with either HSP70 (200 nM), PepFect14 (2µM) or PF14:HSP70 (2µM:200nM). The graphs present the size distribution by intensity
Dynamic light scattering (DLS) revealed another feature of the interactions between PepFect14 and HSP70 (Fig. 1B). A solution containing the complexes was diluted in DMEM + 10% FBS as well as control solutions of HSP70 and PepFect14 alone. The media supplemented with 10% FBS presented two peaks that were due to the serum. Indeed FBS contains a large amount of proteins such as albumins and growth factor that tend to aggregate into nanoparticles. The sample with HSP70 alone showed a similar particle size distribution to the media alone. The reading could not be differentiated from the background indicating that there were no nanoparticle with different size than in FBS formed in this solution. In the case of PepFect14, no new peaks could be detected but a broadening of the second FBS peak was observed. PepFect14 alone might form a particle in the conditions of our experiment but, then, it was also hidden by the FBS background. The sample containing the complexes PF14:HSP70 presented a clear new peak at 712.4 nm. The differences between the size repartition of the complexes and its component alone was an additional proof of the interaction between PepFect14 and HSP70. Furthermore, as usually observed when compounds form a complex with PepFect14, PF14:HSP70 formed nanoparticles. This is of major importance as the ability to form a nanoparticle has been demonstrated to be a key point in transfections mediated by PepFect14 (Lehto et al. 2017).
PepFect14 Mediates HSP70 Transfection
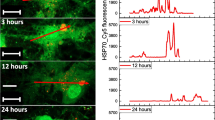
The tryptophan extinction and the DLS results demonstrated that PepFect14 was able to bind to HSP70 and to form a stable complex. It is already known that PepFect14 is able to deliver oligonucleotides when it forms a complex with them. In order to verify that the same properties were kept intact when binding to HSP70, the intracellular uptake was monitored by epi-fluorescent microscopy. HSP70 was labelled with Alexa Fluor 568, before complexation with PepFect14 at molar ratio 3, 5 and 10 and addition to BHM pLuc cells for 1 or 3 h. The cell nuclei were stained using Hoechst 33,258. Control wells where cells were treated by HSP70-Alexa 568 alone were also prepared and imaged. The complexes formed at molar ratio 10 exhibited the best results and the images and corresponding surface plots are displayed in Fig. 2. The cells treated with PF14:HSP70 complexes presented an intense distribution of HSP70-Alexa 568 in their cytoplasm already after 1 h treatment and the red dots repartition became even more intense after 3 h compared to the control cells treated with HSP70-Alexa 568 alone. It must be noted that, in the control cells, HSP70-Alexa 568 could also be detected inside the cytoplasm but in a significantly smaller amount as shown by the images and the surface plots.

Epi fluorescence microscopy. BHM pLuc cells treated with either PF14:HSP70-Alexa568 (2µM:200nM) or HSP70-Alexa 568 (200nM)
The Transfected HSP70 is Biologically Available
To ensure that the delivered HSP70 was available in the cells and able to induce a biological effect, a luciferase activity recovery assay was set up. BHM pLuc cells were treated with lipofectamine 2000 loaded with siRNA against HSPA1A/B to induce a knockdown of HSP70 level before being transfected with PF14:HSP70. First we assessed if our treatment induced a toxic effect that would decrease the cell proliferation and influence our results. A WST-1 assay was performed on the cells. This assay allowed us to monitor the cell proliferation without interfering with the treatment. All our treatment induced a slight decrease in the cell proliferation from around 20% for PF14:HSP70 and Lipofectamine 2000 to 30% for the combined treatment. PF14 alone was not an exception, showing 10% reduction of the cell proliferation (Fig. 3).

Cell proliferation as measured in the WST-1 assay. The graph present the mean ± SEM of 3 technical replicates
On the same plate, the luciferase activity was then measured. The knockdown was expected to increase the amount of misfolded and thus inactive luciferase. Remarkably, The knockdown of HSPA1A/B did not induce a lower luciferase activity as expected but instead an increase by almost 3-fold compared to untreated cells was observed (Fig. 4 A). However, in the cells that were treated with both Lipofectamine 2000 loaded with siRNA against HSPA1A/B and PF14:HSP70, this increase was significantly reduced to only 1.3 fold. When more controls were performed it was noticed that the increase in luciferase activity could be induce by lipofectamine 2000 alone and strangely not with lipofectamine 2000 loaded with a nonsense siRNA (Fig. 4B).

Measurement of the luciferase activity in BHM pLuc cells Treated with lipofectamine 2000 loaded with siRNA against HSPA1A/B on day 1 and PF14:HSP70 on day 2 (A) and controls (B). The data presented are the means ± SEM of 6 technical replicates
The higher luciferase activity could be due to two different causes. Indeed, in the luciferase assay, an increase means a higher quantity of active luciferase in the assay mix. The fact that lipofectamine did not increase the cell growth proved that the increase in luciferase activity by 270% was not caused by a higher number of cells but a higher production of active luciferase. The restoration to 30% above controls achieved by the complex PF14:HSP70 could not be linked to the 30% decrease in cell population compared to untreated cells and was then due to an intracellular effect of the treatment.
Discussion
The tryptophan fluorescence extinction showed a change in the close environment of the protein indicating a non-covalent binding between PepFect14 and HSP70 as suggested by the results exposed in Dowaidar and al26. This binding is further confirmed by dynamic light scattering where a nanoparticle formed by aggregation of the complexes could be detected with an hydrodynamic diameter around 700 nm. Furthermore, the fact that PF14:HSP70 formed nanoparticles, while HSP70 alone did not, indicated that PepFect14 formed a coat around HSP70 that induced the aggregation of the complexes via hydrophobic interactions. This conclusion is further strengthened by the use of molar ratio 10 in the formation of the complex while the maximum extinction of tryptophan fluorescence was already reached at molar ratio 3. The ability to be bound by PepFect14 is of major interest as the first step for transfection using this cell-penetrating peptide is to form a non-covalent complex that can aggregate into nanoparticles (Lehto et al. 2017). To verify that the complexes formed by PepFect14 and HSP70 could be translocated through the plasma membrane, HSP70 was labelled with a fluorophore and its uptake was monitored by epifluorescence microscopy. While a small amount of non-complexed HSP70 could be seen inside the cytoplasm after 1 or 3 h treatment, a significantly higher amount was detected when the cells were treated with PF14:HSP70. It is already known that HSP70 has the ability to get tethered to the plasma membrane from the intracellular side and even to translocate, to some extent, to the extracellular environment (Calderwood et al. 2016; Vega et al. 2008). This ability is usually observed from the inside of the cell to the extracellular environment and is thought to be mediated by exosomes (Calderwood et al. 2016). The uptake of HSP70 inside cells is yet not reported in the literature to our knowledge. Nevertheless, PepFect14 clearly increased the efficacy of the treatment and allowed a larger amount of protein through the plasma membrane. These results, when taken together, showed that besides being able to form a complex with HSP70, PepFect14 mediated the translocation of HSP70 through the plasma membrane to the cytoplasm. A last question raised here is whether HSP70 could be released in the cytoplasm and available for a biological effect or trapped in endosomes and sequestered from signalling pathways. In the luciferase activity recovery assay, the biological activity of the transfected HSP70 was assessed. We expected that the transfection of a siRNA against HSPA1A/B would induce an increased amount of misfolded and unfunctional luciferase that would be monitored as a decrease in the luciferase activity. Furthermore, it was reported in Li et al. (Li et al. 2019) that Lipofectamine 2000 had a direct effect on endoplasmic reticulum unfolded protein response (ER-UPR). This intrinsic effect of Lipofectamine 2000 would then stimulate the production of misfolded luciferase while silencing HSPA1A/B to prevent a self-recovery of the cells. The fact that the siRNA treatment with lipofectamine 2000 induced an increase in the luciferase activity came as a surprise. The cell proliferation assay showed us that the increase was caused by a higher production of luciferase and not by a larger cell population. Furthermore lipofectamine 2000 alone also achieved the same result indicating that the observed effect was not due to a HSPA1A/B knockdown but to an intrinsic effect of lipofectamine. It has been reported that Lipofectamine 2000 treatments had the ability to influence certain gene promoters and modulate global gene expression patterns (Fiszer-Kierzkowska et al. 2011). Notably, in Fiszer-Kierzkowska et al., Lipofectamine 2000 transfections, besides inducing a cellular stress, could influence gene promoters and could trigger the expression of proteins (Fiszer-Kierzkowska et al. 2011). We hypothesized that the promoter of the luciferase reporter gene was activated, directly or indirectly as a response to Lipofectamine 2000 and this activation induced an increase amount of newly produced luciferase. The mechanism behind this increase in luciferase production by cells treated by Lipofectamine 2000 is still unclear but, more interestingly, the cells that were further treated with PF14:HSP70 presented a restoration of the luciferase activity almost to untreated level without causing any significant toxic effect besides Lipofectamine toxicity. This restoration clearly indicated an effect of PF14:HSP70 at the intracellular level and proved once more the uptake of our complexes. Furthermore, the detection of a biological effect led us to the conclusion that HSP70 was released in the cytoplasm and that its active sites were not impeached by PepFect14 coating. Thus, our treatment induced a intracellular biological activity that led to the restoration of a cell process. The exact mechanism of recovery is most likely quite linked to the mechanism of action of Lipofectamine 2000 on the luciferase expression and, thus is still unclear. We hypothesized that the HSP70 degradation pathways were involved. Indeed, HSP70 acts as a chaperone protein that can direct proteins toward several signalling pathways with various finality (Clerico et al. 2015; Deniset and Pierce 2015; Mayer and Bukau 2005). Among these processes can be found autophagy and proteasome activity (Fernández-Fernández et al. 2017; Witkin et al. 2017). The over-expression of luciferase, and possibly other expression regulations, triggered by the lipofectamine 2000 treatment created an imbalance in the cells transcriptome and we hypothesized that the high amount of HSP70 transfected by PepFect14 induced a protection against this abnormal protein level by mediating the transport of luciferase to autophagosome and/or proteasome for recycling. An additional hypothesis on the reduction of the luciferase activity could be the regulation of transcription factor by HSP70. This regulation would be triggered by the high amount of luciferase in the cytoplasm and mediated by HSP70 to reduce the expression of newly produced luciferase. In any case, the level of luciferase was decreased back to untreated level by our treatment and the balance in the proteome was restored. This clear effect, that was not seen when the cells were not pre-treated by lipofectamine, proved that HSP70 was successfully delivered to the cytoplasm and that the PepFect14 coating did not affect its active sites yielding in the addition of active HSP70 in the cells.
Conclusions
In this study, we have experimentally demonstrated that the cell-penetrating peptide PepFect14 transfected the HSP70 protein. First, we demonstrated that PepFect14 was able to non-covalently bind to HSP70 and to form a stable complex that aggregated in nanoparticles. The complexes were then showed to penetrate the plasma membrane and could be detected in the cytoplasm of cells. Finally, we showed that HSP70 in the cytoplasm was able to achieve a protective function by restoring a balance in the proteome after Lipofectamine 2000 induced stress, either through degradative pathways or transcription regulation pathway. These opens a new field of opportunity for PepFect14. Indeed, besides having proved to be able to deliver various sorts of natural or modified oligonucleotides, PepFect14 is now described for the first time as a protein delivery agent. It is also the first occurrence in the literature of a successful delivery of HSP70, to our knowledge. HSP70 is a major protective protein in cells and its transfection could lead to new pharmacologic methods. One of HSP70 role is to protect other proteins and peptide from forming aggregates. Aggregates are involved in many neurodegenerative diseases. In Alzheimer disease, amyloid ß are wrongly processed and aggregates before forming plaques intracellularly and extracellularly. The formation of these plaques leads to neuronal degeneration (Kocahan and Doğan 2017). In Parkinson disease, α-synuclein forms aggregates as well and the aggregation leads to the death of dopaminergic neurons (Dauer and Przedborski 2003). Delivering HSP70 into affected neurons could offer a extra-protection against these peptide aggregations. Furthermore, the delivery of proteins has more straight forward, less aggressive and less permanent features than oligonucleotides transfection. While oligonucleotides transfection can present advantages, protein transfection allows a better modulation of the treatment and does not rely on intracellular transcription machinery. All the results presented in this study makes the transfection of HSP70 mediated by PepFect14 a major advance in the drug delivery field.
References
Andréasson C, Fiaux J, Rampelt H, Mayer MP, Bukau B (2008) Hsp110 is a nucleotide-activated exchange factor for Hsp70. J Biol Chem 283:8877–8884. https://doi.org/10.1074/jbc.M710063200
Bechara C, Sagan S (2013) Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett 587:1693–1702
Bukau B, Deuerling E, Pfund C, Craig EA (2000) Getting newly synthesized proteins into shape. Cell 101:119–122. https://doi.org/10.1016/s0092-8674(00)80806-5
Calderwood SK, Gong J, Murshid A (2016) Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front Immunol 7:159. https://doi.org/10.3389/fimmu.2016.00159
Campanella C, Pace A, Caruso Bavisotto C, Marzullo P, Marino Gammazza A, Buscemi S, Palumbo Piccionello A (2018) Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int J Mol Sci 19. https://doi.org/10.3390/ijms19092603
Clerico EM, Tilitsky JM, Meng W, Gierasch LM (2015) How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol 427:1575–1588. https://doi.org/10.1016/j.jmb.2015.02.004
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909. https://doi.org/10.1016/s0896-6273(03)00568-3
Deniset JF, Pierce GN (2015) Heat Shock Proteins: Mediators of Atherosclerotic Development. Curr Drug Targets 16:816–826. https://doi.org/10.2174/1389450116666150416115423
Dowaidar M, Gestin M, Cerrato CP, Jafferali MH, Margus H, Kivistik PA, Ezzat K, Hallberg E, Pooga M, Hällbrink M, Langel Ü (2017) Role of autophagy in cell-penetrating peptide transfection model. Sci Rep 7:12635. https://doi.org/10.1038/s41598-017-12747-z
Ervin EH, Pook M, Teino I, Kasuk V, Trei A, Pooga M, Maimets T (2019) Targeted gene silencing in human embryonic stem cells using cell-penetrating peptide PepFect 14. Stem Cell Res Ther 10:43. https://doi.org/10.1186/s13287-019-1144-x
Ezzat K, Andaloussi SE, Zaghloul EM, Lehto T, Lindberg S, Moreno PM, Viola JR, Magdy T, Abdo R, Guterstam P, Sillard R, Hammond SM, Wood MJ, Arzumanov AA, Gait MJ, Smith CI, Hällbrink M, Langel Ü (2011) PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res 39:5284–5298. https://doi.org/10.1093/nar/gkr072
Feige U, Polla BS (1994) Hsp70–a multi-gene, multi-structure, multi-function family with potential clinical applications. Experientia 50:979–986. https://doi.org/10.1007/bf01923452
Fernández-Fernández MR, Gragera M, Ochoa-Ibarrola L, Quintana-Gallardo L, Valpuesta JM (2017) Hsp70 - a master regulator in protein degradation. FEBS Lett 591:2648–2660. https://doi.org/10.1002/1873-3468.12751
Fiszer-Kierzkowska A, Vydra N, Wysocka-Wycisk A, Kronekova Z, Jarząb M, Lisowska KM, Krawczyk Z (2011) Liposome-based DNA carriers may induce cellular stress response and change gene expression pattern in transfected cells. BMC Mol Biol 12:27. https://doi.org/10.1186/1471-2199-12-27
Ghisaidoobe AB, Chung SJ (2014) Intrinsic tryptophan fluorescence in the detection and analysis of proteins: a focus on Forster resonance energy transfer techniques. Int J Mol Sci 15:22518–22538
Hartl FU, Hayer-Hartl M (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858. https://doi.org/10.1126/science.1068408
Helmfors H, Eriksson J, Langel Ü (2015) Optimized luciferase assay for cell-penetrating peptide-mediated delivery of short oligonucleotides. Anal Biochem 484:136–142. https://doi.org/10.1016/j.ab.2015.05.023
Kim TK, Eberwine JH (2010) Mammalian cell transfection: the present and the future. Anal Bioanal Chem 397:3173–3178. https://doi.org/10.1007/s00216-010-3821-6
Kim JY, Han Y, Lee JE, Yenari MA (2018) The 70-kDa heat shock protein (Hsp70) as a therapeutic target for stroke. Expert Opin Ther Targets 22:191–199. https://doi.org/10.1080/14728222.2018.1439477
Kocahan S, Doğan Z (2017) Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin Psychopharmacol Neurosci 15:1–8. https://doi.org/10.9758/cpn.2017.15.1.1
Lee YW, Luther DC, Kretzmann JA, Burden A, Jeon T, Zhai S, Rotello VM (2019) Protein Delivery into the Cell Cytosol using Non-Viral Nanocarriers. Theranostics 9:3280–3292. https://doi.org/10.7150/thno.34412
Lehto T, Ezzat K, Wood MJ, El Andaloussi S (2016) Peptides for nucleic acid delivery. Adv Drug Deliv Rev 106:172–182
Lehto T, Vasconcelos L, Margus H, Figueroa R, Pooga M, Hällbrink M, Langel Ü (2017) Saturated Fatty Acid Analogues of Cell-Penetrating Peptide PepFect14: Role of Fatty Acid Modification in Complexation and Delivery of Splice-Correcting Oligonucleotides. Bioconjug Chem 28:782–792
Li Z, Zhang C, Wang Z, Shen J, **ang P, Chen X, Nan J, Lin Y (2019) Lipofectamine 2000/siRNA complexes cause endoplasmic reticulum unfolded protein response in human endothelial cells. J Cell Physiol 234:21166–21181. https://doi.org/10.1002/jcp.28719
Mayer MP, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62:670–684. https://doi.org/10.1007/s00018-004-4464-6
Pinto M, Morange M, Bensaude O (1991) Denaturation of proteins during heat shock. In vivo recovery of solubility and activity of reporter enzymes. J Biol Chem 266:13941–13946
Pisal DS, Kosloski MP, Balu-Iyer SV (2010) Delivery of therapeutic proteins. J Pharm Sci 99:2557–2575. https://doi.org/10.1002/jps.22054
Pratt WB, Toft DO (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 228:111–133. https://doi.org/10.1177/153537020322800201
Qu B, Jia Y, Liu Y, Wang H, Ren G, Wang H (2015) The detection and role of heat shock protein 70 in various nondisease conditions and disease conditions: a literature review. Cell Stress Chaperones 20:885–892. https://doi.org/10.1007/s12192-015-0618-8
Radons J (2016) The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones 21:379–404. https://doi.org/10.1007/s12192-016-0676-6
Rochet JC, Hay BA, Guo M (2012) Molecular insights into Parkinson’s disease. Prog Mol Biol Transl Sci 107:125–188. https://doi.org/10.1016/b978-0-12-385883-2.00011-4
Rokutan K (2000) Role of heat shock proteins in gastric mucosal protection. J Gastroenterol Hepatol 15 Suppl:D12-19. https://doi.org/10.1046/j.1440-1746.2000.02144.x
Sharma D, Masison DC (2009) Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept Lett 16:571–581. https://doi.org/10.2174/092986609788490230
Sindrewicz P, Li X, Yates EA, Turnbull JE, Lian LY, Yu LG (2019) Intrinsic tryptophan fluorescence spectroscopy reliably determines galectin-ligand interactions. Sci Rep 9:11851. https://doi.org/10.1038/s41598-019-47658-8
Strub A, Zufall N, Voos W (2003) The putative helical lid of the Hsp70 peptide-binding domain is required for efficient preprotein translocation into mitochondria. J Mol Biol 334:1087–1099. https://doi.org/10.1016/j.jmb.2003.10.023
Vega VL, Rodríguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, Multhoff G, Arispe N, De Maio A (2008) Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol 180:4299–4307. https://doi.org/10.4049/jimmunol.180.6.4299
Veiman KL, Mäger I, Ezzat K, Margus H, Lehto T, Langel K, Kurrikoff K, Arukuusk P, Suhorutsenko J, Padari K, Pooga M, Langel Ü (2013) PepFect14 peptide vector for efficient gene delivery in cell cultures. Mol Pharm 10:199–210. https://doi.org/10.1021/mp3003557
Vivian JT, Callis PR (2001) Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J 80:2093–2109. https://doi.org/10.1016/s0006-3495(01)76183-8
Witkin SS, Kanninen TT, Sisti G (2017) The Role of Hsp70 in the Regulation of Autophagy in Gametogenesis, Pregnancy, and Parturition. Adv Anat Embryol Cell Biol 222:117–127. https://doi.org/10.1007/978-3-319-51409-3_6
Holmberg MA, Gowda NKC, Andréasson C (2014) A versatile bacterial expression vector designed for single-step cloning of multiple DNA fragments using homologous recombination. Protein Expr Purif 98:38–45. doi: https://doi.org/10.1016/j.pep.2014.03.002
Hageman J, Kam**a HH (2009) Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones 14(1):1–21. doi: https://doi.org/10.1007/s12192-008-0060-2
Acknowledgements
This study was funded through the Swedish Research Council (VR-NT 2017–03691 to ÜL).
Funding
Open access funding provided by Stockholm University.
Author information
Authors and Affiliations
Contributions
MG and ÜL supervised the study, MG designed the experiments, participated in the experiment, analysed the results and wrote the manuscript, LF participated in the experiment and analysis of the result, CPC provided the peptides and participated in the discussion, MC and CA provided the purified protein and participated in the writing of the methods.
Corresponding author
Ethics declarations
Statements and Declarations
None.
Competing Interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gestin, M., Falato, L., Ciccarelli, M. et al. Transfection of Heat Shock Protein 70 kDa (HSP70). Int J Pept Res Ther 28, 105 (2022). https://doi.org/10.1007/s10989-022-10416-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-022-10416-y