
Abstract
A novel method has been developed for the simultaneous determination of difficult-to-measure (DTM) nuclides 93Zr, 237Np, 232Th, 230Th and 228Th using a DGA resin® (containing N,N,N′,N′-tetra-n-octyl-diglycolamide) column for simultaneous separation of Zr and actinides followed by purification of Zr, Np and Th on a TEVA resin® (containing Aliquat 336) column. Samples are destroyed by fusion with sodium hydroxide or by acid destruction using HNO3, HCl and HF. Pre-concentration procedure tailored to the extraction chromatography is based on co-precipitation of Zr and actinides and removal of alumina, silica, iron and calcium. The concentrations of 93Zr, 237Np and Th nuclides are determined by ICP-MS and α spectrometry, respectively. High chemical recoveries (≥ 73%) and separation factors were obtained for Zr, Np and Th both in evaporation concentrates of an NPP and various samples of mineral origin such as soil and concrete. The procedure can be easily combined with the simultaneous separation of other actinides (Pu, Am, U) using the same DGA column that has been described in our previous papers (Cassette et al. in Appl Radiat Isot 68:122–130, 2010 and Groska et al. in J Radioanal Nucl Chem 309(3):1145–1158, 2016).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Zirconium-93, 237Np, and natural isotopes of thorium (232Th, 230Th, 228Th) are regarded as difficult-to-measure (DTM) radionuclides due to the low emission of gamma radiation and their long half-lives.
Zirconium-93 is a long-lived (half-life of 1.61(6)*106·y), pure beta emitting nuclide. It is a fission product and activation product of zirconium used as fuel cladding material in pressurized water reactors. In nuclear waste after approximately 103 years, 93Zr will be the second largest contributor to fission product activity [1]. Because of the long-term safety concerns of nuclear waste disposal, analysis of 93Zr is important. Neptunium-237 is also a long-lived (half-life of 2.144(7)*106·y) nuclide decaying by alpha particle emission. It is a transmutation product in nuclear reactors, and an extinct natural radionuclide. Due to its high mobility it represents a higher safety issue in the environment of nuclear facilities. 228Th (t1/2 = 1.9126(9)·y) belongs to the natural decay series of 232Th (t1/2 = 1.402(6)*1010·y), while 230Th (t1/2 = 7.538(30)*104·y) is a member of the 238U decay series. They are long-lived alpha emitting radionuclides that are present as primordial radionuclides in the environment. 232Th, via the production of the fissile material 233U, is the raw material of the Th based nuclear fuel cycle. 230Th is an important nuclide in geological age determination measuring the 234U/230Th ratio. As DTM nuclides their determination is important both in waste of thorium based fuel cycle and in the environment.
One of the questions that might arise about our concept is the unusual combination of the analytes. Why do we try to combine the determination of these different radionuclides of Zr, Np and Th? Besides the mutual interest to determine DTM nuclides in the nuclear fuel cycle the basic reason is the chemical similarity among Zr belonging to the IVB group of the periodic chart, and the tetravalent actinides, namely Th and Np(IV). Although Np exists in various oxidation states from IV to VI it is easily turned to tetravalent form in reducing media. In our laboratory two combined methods for the simultaneous determination of actinides (Pu, Am–Cm and U isotopes) have been developed, one for the analysis of radioactive waste [2] and one for mineral samples [3]. We planned to extend the procedures for other actinides (Th, Np), and because of the chemical similarity it was evident to include Zr in the separation scheme. Thus an improved method for the analysis of all major and minor actinides and for 93Zr could be obtained allowing the processing of samples with single sample destruction.
A great variety of procedures for the selective separation of major and minor actinides (Pu, U, Th, Np, Am and Cm) and their determination by α spectrometry (AS) or mass spectrometry (MS) have been described in the literature that was summarized in the book by Lehto J. and Hou X. [4]. We have also made a compilation about the references on alpha spectrometry including actinides separations [5].
In our actinide procedure [2], a single extraction chromatographic resin, the DGA containing N,N,N′,N′-tetra-n-octyldiglycol-amide on an inert support [6] was used to retain all actinides from 4 M HCl under reducing conditions. Uranium, Th, Pu and Am–Cm were eluted sequentially after on-column oxidation state adjustments with diluted acids, using complexing agent and changing the temperature of the column and the eluent composition. In the chromatographic separation of model solution high recoveries (above 85%) were obtained without significant cross-contamination (below 5%). The method was successfully applied for the analysis of radioactive waste samples up to 100 mL. Uranium, Pu and Am–Cm alpha sources were counted by alpha spectrometry. It was assumed that in the procedure Th, Np(IV) and Zr behaved similarly and were stripped together.
By improving the procedure a novel method for the analysis of U, Pu, Am nuclides in environmental samples (soil and sediment) has been obtained [3]. Samples were decomposed by fusion and 3 precipitation steps (a mixed hydroxide, a mixed fluoride and a sub-stoichiometric Mg–Ca hydroxide) were included to remove Si, excess of Fe and Ca before the chromatographic separation with DGA. The combined method has been successfully tested for U, Pu, Am nuclides in a set of standard reference materials. The method failed when Th was to be determined.
The aim of the present work was to extend the procedure for the separation of Zr, Th and Np from other actinides and sample components and allow their separation from each other to minimize the interferences during measurement.
93Zr can be detected by inductively coupled plasma mass spectrometry (ICP-MS [7], ICP-MS/MS [8]), liquid scintillation counting (LSC [7]) and accelerator mass spectrometry (AMS [9]). We planned to detect 93Zr by ICP-MS that seemed to be faster, more sensitive and more selective against other radionuclides than LSC. The major interferences in ICP-MS measurements are the isobaric 93Nb (natural abundance is 100%) and stable Zr (containing 92Zr, 94Zr) due to abundance sensitivity. Small amount of radioactive 93Mo in nuclear samples will also cause interference. Therefore chemical separation has to remove Nb and Mo efficiently [10]. We did not consider the use of LSC because we wanted to apply 95Zr yield tracer. 237Np can be detected by alpha spectrometry and ICP-MS. We selected ICP-MS to increase sensitivity. We planned to use 239Np as gamma emitting yield tracer. In this case, chemical separation should provide excellent separation of U to reduce abundance sensitivity due to 238U (nat. abundance in U is 99%). 232Th and 230Th can be well detected by ICP-MS, and by alpha spectrometry. Because 228Th can also be detected by alpha spectrometry this was the method of choice. In this case chemical separation is responsible for removal of matrix and especially Zr to obtain “infinitely” thin alpha source (typically, source thickness should be less than 100 μm).
Separation possibilities of Zr by various extraction chromatographic resins have been studied by a couple of scientists. Traditionally cation and anion exchange resins have been applied, but we favored extraction chromatographic procedures for higher selectivity and speed. Extremely high distribution ratios (Dw > 104) for Zr were measured on Zr resin (containing hydroxamate) in a wide range of HCl and HNO3 concentrations, but Nb was retained similarly strong not offering a simple effective Zr/Nb separation (NPL) [11,12,13]. A procedure was proposed by Russell et al. [13] for the separation of Zr/Nb using 0.01–0.1 M oxalic acid but the conditions have not been optimized.
Very high distribution ratios (Dw > 104) for Zr were measured on DGA resin from HCl (> 1 M) and HNO3 (> 0.01 M) of a wide range of acidity according to Pourmand et al. [14] and a simple Nb separation option was foreseen by changing the acidities.
Dw values of Zr on TRU resin (containing carbamoylmethylphosphine oxide and t-butyl phosphate) [15] from concentrated HNO3 (> 5 M) are also above 104 while those of Nb are about two orders magnitude lower offering a separation possibility according to the measurements of Russell et al. [13].
Oliveira et al. [16] retained Zr on TRU resin from 4 M HCl and eluted with 2 M HCl and TEVA resin from 9 M HCl and eluted with deionized water. Niobium decontamination factors for the whole separation process were ~ 99% for both TRU and TEVA. Shimada et al. [17] also separated Zr from radioactive waste on TRU resin.
It is well known from the historic times on that one of the major interferences in the PUREX uranium, plutonium reprocessing is Zr that is well retained on TBP in a wide range of HNO3 concentrations [18]. The same process takes place on TBP resin although the Dw values are relatively small (between 102 and 103) when HNO3 concentration increases from 1 to 16 M and at the highest concentrations Nb is about an order of magnitude less stronger retained (Dw is about 102) as it was measured by Russell et al. [13].
UTEVA resin (containing dipentylpentyl phosphonate) [19] can also retain Zr both from concentrated HNO3 and HCl. Dw values for Zr and Nb were determined from HCl solutions by Radchenko et al. [19]. They measured Dw > 102 in HCl solutions of higher than 6 M, and found that Nb was even more strongly retained. According to equilibrium studies of Russell et al. the Dw values of Zr on UTEVA from HCl solutions (> 8 M) are also about 103 [13]. We used to determine 93Zr in radioactive waste retaining it from 8 M HNO3 or 9 M HCl and strip** it with 4 M HCl [20]. After a double step separation procedure using two UTEVA columns an average Zr recovery of 65% and a Nb decontamination factor of 103 could be achieved from 50–100 mL evaporation concentrates.
The most frequently used resins for Zr retention were anion exchange resins and their extraction chromatographic analogue, the TEVA resin (containing the quaterner amine Aliquat® 336 [21]). Distribution ratios of Zr and Nb on TEVA from HCl solutions were measured by Russell et al. [13]. Dw values of Zr are fairly low up to a HCl concentration of 10 M, then it rises to > 102. Niobium is much stronger retained on TEVA, Dw values are higher than 103 between 7 and 11 M acidity, therefore a Zr/Nb separation using TEVA from HCl solutions is very promising. Several authors developed methods for the separation of Zr from HCl solutions for various purposes such as geological studies (Ulfbeck et al. [22]), radioactive waste and nuclear material analysis (Oliveira et al. [16] Petrov et al. [23], Shimada and Kameo [17]), application in radiobiology (Radchenko [20]). Remenec et al. [24] studied the retention behavior of Zr and Nb from other solutions (HF, oxalic acid, sulfuric acid) forming strong complexes with Zr and/or Nb. They found that Zr is retained on TEVA from low concentrations of HF (< 0.5 M), sulfuric acid (< 0.25 M) or oxalic acid (< 0.25 M) but these media do not favor a Zr/Nb separation. They performed Zr/Nb separation in a two-step procedure using a cation exchanger for pre-concentration followed by separation on TEVA from 0.5 M HF solution.
Whatever method is selected the separation of Zr, Np and Th is challenging due to special chemical properties of these analytes, i.e. strong tendency for hydrolysis and complex formation. Especially zirconium but to lesser extent Th and Np(IV) are easily hydrolyzed and form sparingly soluble hydroxides and oxides. In low acid concentrations ionic species are not stable. In case of Zr the stability is assured when HCl concentration is > 4 M. At lower concentrations Zr easily adsorbs on the walls of glass beakers. On the other side it is known that most glass types contain Zr as additives responsible for glass hardness and stable Zr can leak from glass into acids interfering with the analysis of stable or radioactive Zr. Furthermore Zr (and to lesser extent Th and Np(IV)) is known to form very stable fluoride complexes, especially in trace concentrations or carrier free form. This is revealed by the strong adsorption of Zr on Teflon surfaces. Determination of Zr and Th nuclides especially in mineral matrices requires complete sample destruction that hardly can be performed by simple acid destruction. Besides HNO3 and HCl the use of HF is unavoidable. Alkaline fusion is a more efficient option for destruction, but evidently the use of Zr crucibles is forbidden. After complete sample destruction a certain amount of fluoride is always present in sample solutions (as a residue of HF reagent or an added HF stabilizer) that is bound to Zr as a complex. A small amount of F− will stabilize dissolved Zr, Th and Np(IV) but the chemical separations have to be optimized taken into account the effect of the fluorides.
We planned to perform a two-step procedure where Zr, Th and Np(IV) are separated from the matrix and other actinides on a DGA resin extending our present procedure for actinides followed by the separation of Zr, Th and Np(IV) individually using a TEVA resin.
Kee** the special properties of Zr, Th and Np(IV) in mind we planned to determine experimentally their Dw values both on DGA and TEVA resins from HCl and HNO3 solutions by batch uptake experiments. Then column separations are to be performed to optimize the chromatographic conditions on both resins. The method is to be tested by analyzing a set of radioactive waste samples spiked with chemical yield tracers. In order to adopt the method for mineral samples the pre-concentration steps (mixed hydroxide, mixed fluoride and sub-stoichiometric Mg–Ca-hydroxide) are to be tested under model conditions. Finally, the method is to be tested with standard reference materials (IAEA-SRM-375, IAEA-SRM-326) and mineral samples (concrete, marl). Performance parameters of the two methods (chemical recoveries, decontamination factors, detection limits) are to be presented.
Experimental
Preparation of radioactive tracers
Carrier-free mixture of 95Zr-95Nb and 239Np tracers was produced by neutron irradiation of natural uranium. 95Zr-95Nb is obtained from thermal neutron fission of 235U while 239Np is obtained from neutron absorption of 238U. After 10 min long irradiation of about 5 mg uranyl nitrate in the Budapest Research Reactor (thermal neutron flux: 2 × 1013 n cm–2 s–1) the sample was dissolved in 5–10 mL 10 M HCl and loaded on a UTEVA column. The column was washed with 20 mL 9 M HCl, and Zr–Nb–Np fraction was stripped with 20 mL 4 M HCl. The strip solution was acidified to 9 M by addition of 12 M HCl and it was purified by repeating the same procedure on a 2nd UTEVA column. The tracer solution was stabilized with traces of HF. 95Zr-95Nb and 239Np activities were determined by gamma spectrometry. Aliquots of this solution were used as radioactive tracers in the tests and during analysis of real samples.
Batch uptake experiments to determine Dw values on DGA and TEVA resins
50–100 mg of DGA resin (50–100 µm particle size) or TEVA resin (50–100 µm particle size) were contacted with 1.5 mL of a load solution spiked with known activities of the radiotracers, i.e. 95Zr, 95Nb, 239Np in PE centrifuge tubes. Solutions contained various concentrations of reagents, i.e. Fe2+ (60–70 ppm), Zr4+ (0–100 ppm), HF traces, H3BO3 (150 ppm).
Samples were shaken for 60 min in a shaker, then settled for 10 min and the phases were separated with a 0.45 µm pore size Nylon syringe filter. Experiments were performed at room temperature (not controlled). The separated liquid phase was counted against standard samples by gamma spectrometer. The distribution coefficient Dw was calculated using the following equation:
where A0 and As are the aqueous phase activity before and after equilibration, w is the weight of the resin in grams, and V is the volume of the aqueous phase in mL (see in reference [6]).
Elution tests to optimize separations on TEVA resin
Column elution tests were performed with spiked samples on TEVA columns (length 40 mm, inner diameter 7 mm). Load sample contained the mixed 95Zr-95Nb and 239Np tracers the activities of which were measured in each fraction and the load by gamma spectrometry. The following elution protocol was used:
-
Condition: 10 mL 12 M HCl
-
Load: 17 mL 11 M HCl containing 0.1–0.5 g Mohr-salt (Fe.2+), 20 μL 70% N2H4
-
Rinse: 10 mL 12 M HCl
-
Zr strip: 10 mL 8 M HCl/ 10 mM HF
-
Rinse (R): 5 mL 8 M HCl/10 mM HF
-
Np strip: 5 mL 4 M HCl
-
Nb strip: 10 mL 7 M HNO3
-
Mo strip: 20 mL 1 M HCl
The test was repeated with 230Th spike (instead of the mixed 95Zr-95Nb and 239Np tracer) that was counted by LSC. The test was repeated with stable Nb and Mo of 0.2 mg each (instead of the mixed 95Zr-95Nb and 239Np tracer) that were measured by ICP-MS.
From the measured amounts of tracers or carriers the % eluted fraction was calculated and plotted as elution chromatograms. From these measurements decontamination factors were calculated.
Tests to co-precipitate Zr and Np and load them on DGA in presence of HF
Co-precipitation with mixed fluoride precipitate
100 mg CaCl2·2H2O (about 25 mg Ca) and 100 mg Fe(NO3)3·9H2O were dissolved in 20 mL 1 M HCl and spiked with the mixed 95Zr-95Nb-239Np tracer. Iron was reduced with 250 μL 70% hydrazine (warmed and cooled) and 5 mL 40% HF were added to make a precipitate. The precipitate was filtered through 0.45 μm pore size (ϕ 25 mm) membrane that was counted by gamma spectrometry, and the retained percentage of 95Zr, 95Nb, 239Np was calculated. The test was repeated when 1 g of Al(NO3)3·9 H2O (about 75 mg Al) was added to the test solution.
Co-precipitation with sub-stoichiometric Ca-Mg hydroxide precipitate
100 mg CaCl2·2H2O (about 25 mg Ca) and 100 mg Fe(NO3)3·9H2O (about 15 mg Fe) were dissolved in 20 mL 1 M HNO3 and spiked with mixed 95Zr-95Nb-239Np tracer. Iron was reduced with 250 μL 70% hydrazine and 62 μL 40% HF were added in equivalent amount with Ca. Then 1.5 g MgCl2·6H2O was dissolved to help sub-stoichiometric precipitation of Ca whith Mg(OH)2. Mg(Ca) hydroxides were precipitated by adjusting the pH to 6 by the addition of 5 M NaOH.
Retention on DGA in presence of HF
A load solution of 16 mL 4 M HCl/0.15 M Na2SO3 was prepared from 8 mL 0.5 M HCl by addition of 95Zr-95Nb tracer, 200 mg Na2SO3 and 8 mL 12 M HCl. HF was added to the load to 10, 50 and 100 mM concentrations. The standard DGA column (0.5 g resin) was loaded and washed with 20 mL 4 M HCl and the combined effluent and wash solution was counted by gamma spectrometry. Yields of Zr and Nb eluted from the column was calculated.
Procedure to separate Zr, Th and Np from radioactive waste and mineral samples (soil, concrete, marl)
For all processes when heating is required, Teflon beakers are used to avoid contamination by stable Zr released from normal glassware. For all processes high purity HF has to be used to avoid sample contamination with stable Zr. Other reagents can be simply of analytical grade.
Acid destruction and load preparation from radioactive waste
Evaporation concentrates from a NPP containing high concentrations of H3BO3 (> 10 g/L) and NaOH (pH > 11) from a few mL up to 100 mL were processed. For spiking the waste samples the purified mixed 95Zr, 95Nb, 239Np tracer of more than 10 Bq activity each, and a 232U/228Th tracer (usually about 0.1 Bq) were added. If Am and Pu isotopes were to be determined then the samples were spiked with 243Am and 242Pu tracers (about 0.1 Bq each).
Destruction of waste samples was done by wet ashing. The waste sample was evaporated to dryness. Evaporation was repeated with about 2*30 mL 65% HNO3, 30 mL 65% HNO3 + 5 mL 30% H2O2, 30 mL 37% HCl. The residue was taken up in 0.5 M HCl (up to 100 mL). Samples were filtered through 0.45 µm pore size membrane. Undissolved residue was transferred in to a Teflon beaker and evaporated with 2–3 times 10 mL 40% HF and finally with 10 mL 37% HCl. The residue was taken up in the necessary amount of 0.5 M HCl and the solution was filtered again through 0.45 µm pore size membrane and combined with the previous filtrate. A solution of maximum 100 mL was obtained.
To prepare a load solution for the DGA column Na2SO3 (2–3 g per 100 mL 0.5 M HCl sample solution) was added to the combined warm filtrate. Reduction of Fe content of the sample was controlled by a Fe(III)-SCN test. If reduction was not complete an extra 1 g of Na2SO3 was added and the test was repeated. When reduction was complete the solution was cooled and 37% HCl was added to it to make it 4 M for HCl. If boric acid crystals appeared in the solution it was further diluted with 4 M HCl as long as the crystals dissolved. The solution was loaded on the DGA column without any pre-concentration, e.g. by precipitation.
Fusion, pre-concentration and load preparation from mineral samples
Fusion with NaOH, dissolution of the melt
A few grams of NaOH flakes are melted at the bottom of a 100 mL nickel crucible. Then 4–5 g of dry, incinerated soil or other mineral sample are weighted in a nickel crucible. It is recommended to use ash to avoid foaming during fusion. Sample amount should be selected according to the Zr content not to overload the columns. The same spikes like in case of radioactive wastes (see 2.5.1) are added in small droplets on the top of the sample and the liquid is evaporated under IR lamp. NaOH flakes are layered on the top (altogether 30 g are used) and the crucible is placed in an ashing oven. The temperature is raised slowly (during about 1 h) up to 550 °C. At this temperature the fusion takes 1 h. Then the crucible is taken from the oven and the sample is cooled.
The solidified melt is dissolved with five 150 mL portions of distilled water that are transferred to 1 L Teflon beaker. The slurry is heated to boiling on a hot plate while stirring intensively. The crucible is rinsed twice with 5 mL 37% HCl and the solution is added carefully to the NaOH solution.
Co-precipitation with mixed (ferrous) hydroxides, dissolution of the mixed precipitates
3 mL of 70% hydrazine and 1 g of Mohr’s salt (Fe(NH4)2(SO4)2·6H2O) dissolved in 20 mL water are added to the solution to obtain in situ ferrous hydroxide precipitate that can co-precipitate all actinides including uranium. The solution is boiled and stirred for an hour, then cooled in water bath to let the precipitate settle. The precipitate is filtered through a polyethersulfone membrane of 0.45 μm pore size and 40 mm diameter. The precipitate is washed with 100 mL 1 M NaOH. The precipitate is transferred from the filter funnel into the original beaker with about 30 mL 37% HCl that is evaporated to dryness. The residue is taken up with 100 mL 1 M HCl. It is heated and stirred.
Co-precipitation using mixed fluorides, dissolution of the precipitate
About 2 mL 70% hydrazine are added to reduce iron. If necessary further 1 mL portions of hydrazine and 25% NH3 are added to complete the reduction of iron indicated by a negative Fe(III)-thiocyanate test. 1 g of CaCl2·2H2O are dissolved then the solution is cooled to room temperature in water bath and 25 mL 40% HF are added that is stirred for one hour. Fluoride precipitates are filtered through a cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter (the mass of which was determined in advance) using a plastic funnel. The precipitate is washed with 20 mL 1:5 HF and filtered again. The precipitate is dried under IR lamp until constant weight and its mass is measured by weighting.
The precipitate is transferred with 45 mL 3 M HNO3 to a Teflon beaker that contains boric acid. The amount of boric acid is calculated from the estimated mass of the fluoride precipitate assuming that the precipitate consists of exclusively CaF2. The mass of boric acid is equimolar compared to the calculated moles of fluoride.
The sample is heated to boiling for 10 min. Additional amount of boric acid (up to 1 g) and a few mL of 65% HNO3 may be added if the sample does not seem to be dissolved. The solution that might be slightly opaque is diluted with distilled water to 150 mL (it becomes about 1 M).
Co-precipitation using mixed (calcium-magnesium) hydroxides, dissolution of the precipitate
0.5 g of Fe(NO3)3·9H2O and 2 mL 70% hydrazine are added to the hot solution that is heated and stirred as long as Fe(III) is reduced to Fe(II) as indicated by a negative thiocyanate test. If necessary few more drops of hydrazine can be added. 11.25 g of MgCl2·6H2O are added and dissolved. The solution is cooled to room temperature in water bath. The pH of the solution is adjusted to 6 by the addition of 5 M NaOH when greenish-brownish colored precipitate is formed. The suspension is stirred for further one hour, then centrifuged and filtered through cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter.
The precipitates are combined and transferred with 20 mL 9 M HCl to a Teflon beaker. About 30 μL 40% HF are added and the solution is stirred for 15 min. The solution is evaporated to dryness, evaporation is repeated with 2 mL 65% HNO3 then with 5 mL 37% HCl and with 2 g of H3BO3 and 5 mL 37% HCl. The residue is dissolved in 20 mL 1 M HCl, finally diluted with 20 mL distilled water to obtain 40 mL 0.5 M HCl solution.
Load preparation
1 g of Na2SO3 and 0.5 g of Mohr’s salt are added to the solution to reduce actinides. The solution is warmed and stirred for 10 min and the reduction of Fe is checked by a negative thiocyanate test. (The solution is not necessarily clear during this step.) Further amount of boric acid might be added to the solution to help dissolution, but without the appearance of undissolved crystals. The solution is cooled to room temperature in water bath and 20 mL 37% HCl are added to adjust acidity to about 4 M. The solution is filtered through cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter. (If filtration is slow centrifuging may precede the filtration).
Between the strip solutions of U, Th–Np–Zr, Pu and Am-Cm the column is rinsed, and occasionally oxidation states of actinides are adjusted to reduce cross-contamination. These fractions are rejected.
Chromatographic separation of Zr, Th and Np from matrix on DGA resin column
The chromatographic separation procedure is applied in the same way for the load solutions obtained by “Acid destruction and load preparation of radioactive wastes” and by „Fusion, pre-concentration and load preparation of mineral samples” according to Experimental Sects. “Acid destruction and load preparation from radioactive waste” and “Fusion, pre-concentration and load preparation from mineral samples”, respectively. Typically 60–150 mL of load solution is processed.
Column preparation: 0.5 g DGA resin® (registered trade mark of TRISKEM International) of 50–100 μm particle size was soaked in 4 M HCl for a couple of hours, it was packed into PE chromatographic column with 7 mm inner diameter. Quartz sand was used for the top bed support against clogging.
Separation on DGA
The column of DGA resin was preconditioned with 10 mL 4 M HCl solution. For temperature control jacketed columns were prepared by surrounding the standard PE columns with home-made glass jackets and circulating water from a standard temperature control unit. The temperature of the eluents was also controlled using the water bath system of the same unit. After loading the column the elution sequence according to Table 1 was followed.
Chromatographic separation of Zr, Th, Np on TEVA resin column
Th, Np(IV) and Zr from the common Th, Np(IV), Zr strip solution are further separated on a TEVA resin column. From U, Pu, Am strip solutions alpha sources can be prepared directly (U, Am) or after a simple pre-treatment (Pu) by micro-co-precipitation according to [1, 2].
Processing of Zr, Np, Th strip solution from DGA
The Th, Np(IV), Zr strip solution is evaporated in a small Teflon beaker to dryness, then it is repeatedly evaporated to dryness with 2 mL 65% HNO3 + 1 mL 37% HCl, 2 mL 37% HCl. The residue is taken up with 2 mL 4 M HCl, gently warmed with 20 μL 70% N2H4, then cooled to room temperature. It is diluted with 15 mL 12 M HCl to obtain about 11 M concentration.
Separation of Zr, Np, Th on TEVA
Column preparation: TEVA resin® (registered trade mark of TRISKEM International) of 50–100 μm particle size was soaked in distilled water, it was packed into PE chromatographic column with 7 mm inner diameter up to 40 mm height and covered with quartz sand.
The TEVA column is conditioned with 10 mL 12 M HCl, then the sample solution is loaded on the column and washed with 10 mL 12 M HCl. Effluent and rinse are collected to prepare Th source. Zirconium is stripped with 10 mL 8 M HCl/10 mM HF. The column is rinsed with a further 5 mL 8 M HCl/ 10 mM HF. Finally Np is stripped with 5 mL 4 M HCl.
Source preparation for alpha spectrometry and ICP-MS
Micro-co-precipitation is used for Th α source preparation as follows:
The Th strip solution is evaporated to 1–2 mL, than diluted with 20 mL 0.1 M HCl. A solution containing 50 μg Nd as Nd(NO3)3 and 5 mL 40% HF are added. After half an hour, the micro-precipitate is filtered slowly through a membrane (e.g., polyethersulfone) of 0.10 μm pore size and 25 mm diameter. Filters are dried under IR lamp.
The Zr and Np strip solutions are collected in special Teflon beakers that have a conical shape and can be gently evaporated to a single droplet. The residue is evaporated once more with a mixture of 1–1 drops of 65% HNO3, 37% HCl, 40% HF. The residue is taken up with 10 mL 0.1 M HCl/10 mM HF.
Measurements
Alpha spectrometry (AS)
Passivated Ion Implanted Si detectors (AMETEK, type TU-019-300-AS) attached to EG&G Ortec 576A dual alpha spectrometer and EG&G Ortec Ethernim multichannel analyzer were used to collect alpha spectra. Sources were measured in evacuated chambers at an approximate source-detector distance of 5 mm and source diameter of 25 mm. EG&G Ortec Maestro emulation software was used to evaluate the spectra. Chemical recoveries relative to the activities of the tracers added and the actinides of the analytes according to isotope dilution were calculated.
Gamma spectrometry
HPGe detector (Canberra No.: 7229P) attached to a Canberra multichannel analyzer (9615 Amplifier, 9645 HV supply unit, 9633 ADC and 556 AIM) was used to collect gamma spectra of 95Zr, 95Nb and 239Np tracer. Canberra Genie-2000 software package was used to evaluate the spectra. Chemical recoveries relative to the activities of the tracers added were calculated.
Liquid scintillation counting (LSC)
A Perkin Elmer TriCarb 2800 facility was used to measure the samples of the column elution test containing 230Th. Eluted percentage was calculated against the added activity of the tracer (A0).
Microwave plasma atomic emission spectrometry
(MP-AES): An Agilent MP-AES 4200 spectrometer was used to measure total Zr in the Zr- and Np-strip solutions, in order to determine the dilution of samples required for ICP-MS measurements. Calibration solutions were prepared from a mono-element standard stock solution.
Inductively coupled plasma mass spectrometry (ICP-MS)
An Agilent Technologies 7500a ICP-MS was used to measure Zr isotopes (natural and 93Zr), 93Nb, Mo isotopes and 237Np. Calibration solutions were prepared from mono-element standard stock solutions of Zr, Nb and Mo and and 237Np. Calibration solutions were prepared with high-purity water and suprapure HCl. Calibration curve for 93Zr was calculated by averaging the sensitivity of 92Zr and 94Zr.
Results and discussion
A novel chromatographic procedure has been developed at our laboratory and published recently for the simultaneous separation of all actinides (U, Th–Np, Pu, Am–Cm) from radioactive waste [1] and mineral samples [2] using a single small DGA resin column. The procedure for waste analysis consists of acid destruction including HF destruction of the undissolved residue, separation of actinides (U, Pu, Th, Am-Cm) on a single DGA column where loading solution obtained from 20–100 mL evaporation concentrate is about 75–150 mL 4 M HCl containing 0.15 M Na2SO3 reducing agent. The species that are retained under these conditions are U(IV), Pu(III), Th(IV), Am(III). Actinides are sequentially eluted by changing the eluents, their concentrations, the oxidation states of actinides and forming complexes. After removal of non-retained elements such as Ca and Fe, uranium is the first actinide that is stripped with dilute HNO3 (0.5 M). Then Pu is on-column reduced to Pu(III) with Fe(II), and the tetravalent actinides are stripped selectively with oxalic acid in 0.5 M HNO3. After oxidizing Pu back to Pu(IV-VI) with the strong oxidizing agent K2S2O8 it is stripped with oxalic acid in 0.5 M HNO3. Am is stripped at the end with 0.5 M HCl. The exact conditions of the actinide separation are summarized in Table 1. In this procedure there is no need for pre-concentration of actinides, but it can be performed by co-precipitation on ferrous hydroxide, thus reducing the volume of the load down to 20 mL. With this procedure good chemical recoveries and high decontamination factors for the whole procedure were obtained (Table 2).
Later we extended the procedure for the determination of actinides from soil and sediment [2]. To speed up the long destruction 4–5 g of samples were destroyed with NaOH fusion. A three-step pre-concentration procedure was developed to remove the major matrix components, i.e. silica, excess amount of Al, Fe and Ca. Actinides were pre-concentrated on i) the mixed hydroxide precipitate obtained by dissolution of the NaOH melt, ii) mixed fluoride precipitate formed by addition of HF, iii) sub-stoichiometric Ca-Mg hydroxide. Conditions of pre-concentration were optimized. After pre-concentration a load solution was prepared with the same composition as in case of waste samples (60 mL 4 M HCl/0.15 M Na2SO3) and the same chromatographic procedure was performed followed by alpha source preparation.
The average recoveries of a model solution on a DGA column, those of a whole analysis procedure from 20–100 mL of evaporation concentrate of an NPP and from 4–5 g of soil and sediment samples are shown in Table 2.
High chemical recoveries were measured for U, Pu and Am, but the procedure failed when Th was analyzed in environmental samples. (In wastes there was no interest to detect Th isotopes.) We did not know the exact reason for failure, however, hydrolysis of Th and adsorption on vessel walls was suspected. Furthermore we realized that the Th strip solution contained Np and Zr. With this observations our goal was to improve the present method likely to extend it for the determination of Th, Np and Zr isotopes.
To understand the theoretical background we decided to perform batch uptake experiments on DGA resin.
Batch uptake experiments
Although there are a few data on the distribution of Zr and Nb on DGA (see Introduction), we decided to perform measurements on Zr, Nb and Np under our well defined experimental conditions.
Tests were performed both in HCl and HNO3 media. The mixed 95Zr-95Nb-239Np tracer was used and Zr carrier was added to represent the expected conditions when mineral samples which usually contain stable Zr are processed. Because the tracer solution always contained some HF we also added boric acid to complex excess F−. Mohr salt was also added to the load (originally 0.5 M HCl) to reduce Np to Np(IV), and acidy was adjusted before loading. We did not measure the Dw values of Th but used published data [5]. Experimental conditions are described in Sect. 1.2. Calculated results are shown in Table 3.
We repeated a few tests without the addition of stable Zr and results were not reproducible. We also confirmed that increasing the HF concentration without H3BO3 Dw values of Zr, Np and Nb were reduced and we found that exact Dw values depend on oxidation state adjustment, HF and Zr carrier concentrations. The basic conclusion of the measurements is that Th, Np(IV) and Zr are strongly retained (Dw > 1000) on DGA if HCl concentration is ≥ 4 M, or HNO3 concentration is ≥ 3 M, and there is chance for good separation of Nb. Because in our method for actinides separations the load solution is 4 M HCl this seems a good solution for extension of the method.
Based on the literature review we planned to separate Th, Np, Zr from each other on a TEVA column. Therefore we performed batch uptake experiments similarly with TEVA resin. Results are summarized in Table 4. We did not measure the Dw values of Th but used published data of Horwitz et al. [19], and also added published data on Np(IV) retention by Horwitz et al. [19].
The conclusion can be drawn that Np(IV), Zr as well as Nb can be retained on TEVA from highly concentrated HCl (11 M). Zr and Nb are not retained from HNO3.The Dw values of Np(IV) measured by us are lower than those of Horwitz et al. but the differences in 9 M HCl can be interpreted in terms of different conditions. The Dw values of Np from 8 M HNO3 differ significantly. Probably the Np(IV) oxidation state was not attained by the addition of Fe2+ that itself is not stable at higher HNO3 concentrations and HF traces can inversely affect the retention. We planned to perform the Th, Np, Zr, Nb separation on TEVA column from HCl solution. To optimize the conditions column separation tests were performed as follows.
Elution tests to optimize separations on TEVA resin
To optimize the conditions, column separation tests were performed. Assuming that the Th–Zr–Np strip solution from DGA resin contains Nb, and optionally Mo, column separation tests were performed with 95Zr-95Nb-239Np tracer, with 230Th spike and Mo and Nb carriers. 95Zr, 95Nb and 239Np were measured by gamma spectrometry, 230Th was measured by LSC, Mo and Nb carriers were measured by ICP-MS.
To obtain the possibly highest HCl concentration, the DGA strip solution was evaporated to dryness. The presence of Mohr-salt and especially sulfate ions helped to evaporate Zr–Nb without adsorption on Teflon surfaces. The residue was taken up in 2 mL 4 M HCl where the reduction of Np to Np(IV) with hydrazine was possible. By “dilution” with 12 M HCl, finally, a 11 M load solution was prepared. Th was released in the effluent, Zr was selectively stripped with 8 M HCl/10 mM HF, finally, Np was eluted with 4 M HCl. The elution chromatograms of Th, Zr, Nb, Np and carrier Nb, Mo on TEVA column are shown in Figs. 1 and 2.

Elution chromatograms of Th, Zr, Nb and Np on TEVA column. Load: 15 mL 11M HCl + Rinse: 5 mL 12M HCl, Zr strip: 20 mL 8M HCl/10mM HF, Np strip: 20 mL 4M HCl

Elution chromatograms of Nb and Mo on TEVA column. Load: 17 mL 11M HCl, Rinse: 10 mL 12M HCl, Zr strip: 10 mL 8M HCl/10mM HF, Zr rinse (R): 5 mL 8M HCl/10mM HF, Np strip: 5 mL 4M HCl, Nb strip: 10 mL 7M HNO3, Mo strip: 20 mL 1M HCl
According to the chromatogram in Fig. 1 with slight modification of the eluent volumes—an optimized separation protocol was developed that is described in Sect. 1.5.4. By increasing the Zr rinse solution to 10 mL, decreasing the Zr and Np strip solutions to 10 and 5 mL, respectively, a good separation of Th, Zr and Np was achieved while chemical recoveries were high (> 90%). Nb contaminates the Np fraction but it does not mean an issue for ICP-MS measurement of Np.
According to the chromatogram in Fig. 2 Np fraction contains most of Nb (69%) and the so called Nb fraction contains all Mo with some Nb. It was calculated that the decontamination factors of Zr from Nb and Mo during the TEVA separation are about 7000 and 900, respectively. Chromatographic tests on DGA resin with Nb and Mo were also performed and decontamination factors of Zr from Nb and Mo were 4 and 35, respectively. This means a final decontamination factors for the whole procedure of 28,000 (Nb) and 31,500 (Mo).
Determination of 93Zr, 237Np and Th isotopes in radioactive wastes
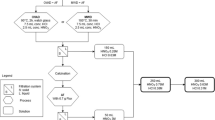
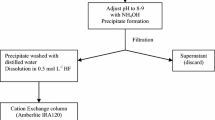
Based on our previous experience in actinide analysis and the recent results of the tests a relatively simple and efficient separation scheme was developed for the determination of 93Zr, 237Np and Th isotopes in radioactive wastes. The flowchart of the procedure is shown in Fig. 3 (left side), the exact experimental conditions are described in Sect. 1.5.1, 1.5.3 and 1.5.4.

Flowchart of separation of Zr, Np and Th (and other actinides) from radioactive wastes (left side) and mineral samples (right side)
The procedure consists of acid destruction, load preparation, chromatographic separation on DGA and TEVA columns. Because of the Dw values for all analytes on DGA are high there is no need for a pre-concentration. Even 150 mL solution can be loaded on a small (0.5 g) DGA column and the risk of adsorption losses on precipitates, especially those of Zr and Nb can be eliminated, thus significantly increasing the overall chemical recoveries.
Analyzing 10 evaporation concentrates from an NPP the following improved overall chemical recoveries were obtained: Zr: 90% ± 4%; Np: 86% ± 9%; Th: 73% ± 54%.
It has to be mentioned that theoretically the chemical recoveries of actinides have not been changed from the values given in the 2nd column of Table 1, but to obtain accurate results for U and Pu isotopes the 95Zr-95Nb-239Np tracer has to be extremely well purified and its purity has to be checked. Typically 2 step purification with UTEVA resin is not enough to measure activities below 1 Bq/L.
The detection limit of 93Zr in evaporation concentrates was about 2–3 Bq/L and the limiting factor in the minimum detectable activity is the stable Zr content of the sample. In our samples it was about 10–20 ppb. If Zr concentration is much higher (e.g. > 1 ppm), the ICP-MS sample has to be diluted by 100 or 1000 times in order to avoid memory effects and instrument failure. Sample dilution means the detection limits are also increased by 100 or 1000 times. Fortunately this is a rare case in waste samples unless Zr fuel cladding damaged.
The detection limit of 237Np in evaporation concentrates was about 2–5 mBq/L. In some samples 10 times higher activity concentrations were detected. The detection limit of 237Np determination by ICP-MS was significantly improved when Np and Zr were separated and there was no need for dilution of Np containing sources.
The detection limit for the alpha emitting 232Th and 230Th isotopes is 40–80 mBq/L, but radioactive wastes typically do not contain Th isotopes, and usually there is no need for their determination.
Tests to co-precipitate Zr and Np with different precipitates
To determine the actinides from soil and mineral samples the procedure for radioactive waste samples had to be modified (see ref [2]). To speed up the destruction alkaline fusion with NaOH was performed (see in Sect. 1.5.2). Silica, bigger amounts of Fe and Ca present in mineral samples in contrast to wastes had to be removed before chromatography on DGA. For that purpose three pre-concentration steps were included between sample destruction and load preparation for DGA, i.e. mixed hydroxide precipitate obtained during dissolution of the fusion cake, mixed fluoride precipitate to remove Si and mixed Ca-Mg hydroxide to reduce the Ca amount. We had to test how Zr–Nb and Np (optionally Th) behave in these precipitation or co-precipitation processes. Because Th radionuclides are DTM alpha emitters therefore we focused our tests on gamma emitting Zr, Nb and Np nuclides.
Mixed hydroxide precipitate
By dissolving the fusion cake with distilled water a strong basic NaOH solution is formed where Si and Al are in dissolved form and are removed by filtration from the mixed hydroxide precipitate that including ferrous hydroxide responsible for adsorption of all actinides in their reduced forms (III or IV) as well as Zr and Nb hydroxides quantitatively.
Mixed fluoride precipitate
Tests were performed with model solutions to check the conditions when Zr and Np are co-precipitated with Ca fluoride. Model solutions were prepared to simulate the real condition in a 1:5 scale when mixed fluoride precipitates are formed. Mixed 95Zr-95Nb-239Np tracer was used to spike the solution. Experimental conditions are described in Sect. 1.4.1, results are shown in Table 5.
It was confirmed (test 1) that Np and Zr are quantitatively co-precipitated with CaF2 while Nb remains in the filtrate as fluoride complex. Good recoveries for Zr and Np are obtained when the precipitate is formed at room temperature favoring the formation of microcrystals and during short contact time (stirring for not longer than 1 h). If the agitation is longer and done at higher temperature Np oxidation state is increased and Np losses during co-precipitation occur, at the same time Nb co-precipitation is increased to spoil Zr/Nb separation.
It was observed (test 2a) that in presence of Al co-absorption of Zr is spoilt (recovery is reduced from 97 to 12%). This effect can be partially compensated by the addition of more Ca, thus obtaining altogether 69% recovery of Zr (test 2b).
Sub-stoichiometric Ca hydroxide precipitate in presence of Mg hydroxide
Tests were performed with model solutions to check the conditions when Zr and Np are co-precipitated with Mg(Ca) hydroxide. Model solutions were prepared to simulate the real condition in a 1:5 scale. It was also simulated that Ca in the solution originated from a previous CaF2 precipitate, therefore equivalent amount of F− was added to the solution as HF. Mixed 95Zr-95Nb-239Np tracer was used to spike the test solution. Experimental conditions are described in Sect. 1.4.2, results are shown in Table 6.
From tests 1 it is clear that co-precipitation of Np(IV) and Zr is limited when free F− is present in equivalent amount with Ca. After longer agitation time recoveries become even poorer (test 1b). High recoveries both for Np and Zr are obtained when precipitation is formed after boiling the solution with an excess of H3BO3 (4:1 mol ratio) and cooling down the solution to room temperature before pH adjustment to 6 (test 2a), and the precipitate is stirred for a short time (1 h). When agitation time is prolonged to 24 h (test 2c) recoveries of Np and Zr are reduced to 55 and 14%, respectively, probably to the back-oxidation of Np to Np(V) and back-extraction of F− ions from H3BO3 to the re-dissolved Zr.
Retention on DGA in presence of HF
Tests were performed to determine the effect of free F− ions on the retention of Zr and Nb on DGA. Test conditions are described in detail in Sect. 1.4.3 and results are shown in Table 7.
High retention of Zr on DGA (97%) was obtained when HF concentration was ≤ 10 mM. Advantageosly, Nb is much less retained (22%) under the same conditions. When HF concentration in the load is increased Zr is less retained similarly to Nb. The 100 mM HF concentration can be compensated with boiling the load with an excess of H3BO3 (5:1 mol ratio), the retention of Zr becomes 100%, but that of Nb is also improved (69%). It is recommended to keep the HF concentration in the load around 10 mM. Other experiments also confirmed that by the addition of AlCl3 to the load the effect of F− ions can also be compensated.
Determination of 93Zr, 237Np and Th isotopes in mineral samples
According to the results of the tests (Sect. “Tests to co-precipitate Zr and Np and load them on DGA in presence of HF”) we built up a modified separation scheme that is adequate for the separation of all actinides and Zr. The procedure consists of destruction of 4–5 g of soil, sediment or other mineral samples with NaOH fusion. Actinides and Zr are pre-concentrated on three precipitates, a mixed hydroxide formed in the dissolution of the fusion cake, a mixed fluoride formed by the addition of HF, and a sub-stoichiometric Mg(Ca) hydroxide (Sect. 1.5.2). The residue is loaded on a DGA column where U, Th-Zr-Np, Pu and Am-Cm are consecutively stripped (Sect. 1.5.3). Th, Zr and Np are finely separated on a TEVA column (Sect. 1.5.4). The whole procedure is described in Sects. 1.5.2–1.5.4 and the flow chart is shown in Fig. 3 (right side).
The major modifications to the original procedure for U, Pu, Am determination [3] in mineral samples that allowed the separation of Th, Np and Zr are the following:
-
For fusion nickel crucible, for chemical treatment Teflon (plastic) equipment has to be used during the whole procedure to avoid Zr releases from glassware.
-
HF has to be used in many operations to avoid Zr adsorption on walls. HF should be supra pure to lower Zr contamination. Zr-F complexes can be decomposed with the addition of boric acid in excess to equimolar amounts and by boiling in acid solution. (Aluminum salts can play similar role in the processes.)
-
Mineral samples have to be decomposed by NaOH fusion where aluminum and silica are removed by dissolution of the fusion cake, otherwise Al would interfere with fluoride co-precipitation of Zr.
-
Co-precipitation with mixed fluorides will remove not only Si but Al remains. It has to be performed from cold solution with max one hour of agitation in the absence of Al. Small amount of Al can be compensated with the addition of more Ca. It is evident that the procedure will not work when Al is not removed like in case of acid destruction. Fluoride precipitate must be dried in order to remove excess HF.
-
Co-precipitation with sub-stoichiometric Mg(Ca) hydroxide has to be performed from cold solution with max one hour of agitation in the absence of F− ions. Fluoride ions are bound as B-F complexes by addition of excess H3BO3.
-
Strong retention on DGA column is assured if F− ions are complexed with addition of excess H3BO3 to the 4 M HCl/0.15 M Na2SO3 load solution.
-
Th, Zr, Np are separated from each other and Zr is separated from Nb and Mo on a TEVA column where sample is loaded from 11 M HCl, Th gets in the effluent and rinse, Zr is stripped with 8 M HCl/10 mM HF, Np is stripped with 4 M HCl.
-
Th isotopes are measured by alpha spectrometry, while 93Zr and 237Np are measured by ICP-MS.
The novel procedure has been tested/applied by the measurement of two standard reference materials; i.e., IAEA-SRM-326 and IAEA-SRM-375 and a standard marl (CT CEB 2297-80) sample and a common concrete (candidate material for application in decommissioning). Good chemical recoveries were obtained for Th, Zr and Np for the whole procedure starting from 4 g of dry material or ash: Th: 97% ± 6%; Zr: 78% ± 13%; Np: 79% ± 20%.
Results of Th analysis obtained by alpha spectrometry are summarized in Table 8. Unfortunately, reference values are only available for 232Th and 230Th in the two SRM materials.
Acceptable results were obtained in the reference materials for Th isotopes. Relative bias of the results are ≤ 12%, z-score values are ≤ 2.1. Z-score criteria is ≤ 2.
Results of 237Np and 93Zr analyses are summarized in Table 9.
Amounts/activities of 237Np, 93Zr were determined by ICP-MS and all the measured values were below detection limits (LD). Detection limits for 237Np are acceptably low (below 1 Bq/kg), but the LD values of 93Zr are high and the reason for these high values is that the stable Zr content of the samples was high (up to 390 ppm) and the samples had to be diluted 500–1000 times before measurement thus increasing the LD values by 500–1000 times, as well. The measured stable Zr content and also the reference values are shown in the table. The good agreement confirms that the chemical separation worked very well for Zr. The abundance sensitivity of the instrument used in this study was in the 10–4 range for 92/93 and for 237/238 mass pairs. To obtain lower detection limits other measuring technique has to be applied for 93Zr determination. LSC could be a good choice but in this case radioactive 95Zr tracer should not be used for recovery determination. Another option is the use of the more sensitive AMS technique [9] where abundance sensitivity is negligible.
Conclusions
A novel procedure for the separation and determination of 93Zr, 237Np, 232Th, 230Th and 228Th has been developed for radioactive waste and mineral samples by testing and modifying the procedures previously developed for the determination of U, Pu and Am-Cm nuclides. The method is based on simultaneous separation of actinides and Zr on DGA resin column followed by separation and purification of Zr, Np and Th on TEVA resin column. Thorium radionuclides are measured by alpha spectrometry, 237Np and 93Zr by ICP-MS. High chemical recoveries were obtained for all analytes and in both sample types in the whole procedure (≥ 73%). The method was tested by the analysis of SRMs and acceptable good agreement was found with the reference values of Th radionuclides and stable Zr. Detection limits were acceptable low for Th and Np nuclides but high for 93Zr nuclide because of the high abundance sensitivity of ICP-MS measurement due to high stable Zr content of the mineral samples. The method was successfully applied for the analysis of radioactive waste samples.
Data availability
The data that support the findings of this study are available from the corresponding author Istvan Papp, upon reasonable request.
References
Cassette P, Chartier F, Isnard H, Fréchou C, Laszak I, Degros JP, Bé MM, Lépy MC, Tartes I (2010) Determination of 93Zr decay scheme and half-life. Appl Radiat Isot 68(2010):122–130
Groska J, Vajda N, Zs M, Bokori E, Szeredy P, Zagyvai M (2016) Determination of actinides in radioactive waste after separation on a single DGA resin column. J Radioanal Nucl Chem 309(3):1145–1158
Vajda N, Zagyvai M, Groska J, Zs M, Bokori E, Braun M (2020) Determination of uranium, plutonium and americium in soil and sediment by a sequential separation procedure using a single DGA column. J Radioanal Nucl Chem 326:695–710
Lehto J, Hou X (2011) Chemistry and analysis of radionuclides. Wiley, Weinheim
Vajda N, Pöllanen R, Martin P, Kim C-K (2018) Alpha Spectrometry. Chapter V. In: L’Annunziata MF (ed) Handbook of Radioactivity Analysis (Fourth Edition). Elsevier Science B.V, Amsterdam, pp 363–422
Horwitz EP, McAlister DR, Bond AH Jr, Barrans RE (2005) Novel extraction chromatographic resins based on tetraalkyldiglycolamides: characterization and potential applications. Solvent Extr Ion Exch 23:319–344
Osváth S, Vajda N, Zs M, Kovács-Széles E, Braun M, Halász M (2017) Determination of 93Zr in nuclear power plant wastes. J Radioanal Nucl Chem 314:31–38
Russell BC, Warwick PE, Mohamud H, Pearson O, Yu Y, Thompkins H, Goddard SL, Croudace IW, Zacharauskas Z (2023) Development of a single method for direct measurement of multiple radionuclides using ICP-MS/MS. J Anal At Spectrom 38:97–110
Pavetich S, Wallner A, Harry Bottero L, Fifield K, Froehlich MB, Huang Y, Koll D, Révay Z, Slavkovská Z, Sterba JH, Tims SG (2022) Accelerator mass spectrometry measurements of 93Zr for astrophysical and nuclear technology applications. Nucl Instrum Methods Phys Res Sect B Beam Interact Mater Atoms 527:45–51
Osváth SZ (2022) Private communicationOsváth S, Hou X, Roos P, Qiao J, Markovic N (2018) Determination of 93Mo (and 94Nb) in nuclear decommissioning waste from a nuclear reactor. Sound/Visual production (digital) https://backend.orbit.dtu.dk/ws/portalfiles/portal/161808178/RadChem2018_93Mo_verzML_v13_1_.pdf
Basic data on ZR resin: TRISKEM International homepage: https://www.triskem-international.com/ resins-and-accessories.php: PS_ZR-Resin_EN_210908.pdf
Dirks C et al (2015) On the development and characterisation of an hydroxamate based extraction chromatographic resin. In: Presented at the 61st RRMC, October 25th–30th, 2015, Iowa City http://www.rrmc.info/rrmc-61/rrmc-61-062a.pdf
Russell B, Ivanov P, Thompkins H, Jerome S (2018) Extraction chromatography separation of zirconium-93. https://www.eichrom.com/wp-content/uploads/2018/07/06.pdf
Pourmand A, Dauphas N (2010) Distribution coefficients of 60 elements on TODGA resin: application to Ca, Lu, Hf U and Th isotope geochemistry. Talanta 81(3):741–753
Horwitz P, Chiarizia R, Dietz M, Diamond H, Nelson D (1993) Anal Chim Acta 281:361–372
Oliveira TC, Monteiro RPG, Kastner GF, Bessueille-Barbier F, Oliveira AH (2014) Radiochemical methodologies applied to determination of zirconium isotopes in low-level waste samples from nuclear power plants. J Radioanal Nucl Chem 2014(302):41–47
Asako S, Yutaka K (2017) Separation of Zr in the rubble waste generated at the Fukushima Daiichi Nuclear Power Station. J Radioanal Nucl Chem 311:1613–1618
Geier RG (1979) Purex process solvent literature review RHO-LD-74 informal report. https://www.osti.gov/servlets/purl/5650046
Horwitz P, Dietz M, Chiarizia R, Diamond H (1992) Analytica Chimica Acta. 266: 25–37; Eichrom Reference HP392. (2) Adriens AG, Fassett JD, Kell
Radchenko V, Filosofov DV, Bochko OK, Lebedev NA, Rakhimov AV, Hauser H, Eisenhut M, Aksenov NV, Bozhikov GA, Ponsard B, Roesch F (2014) Separation of 90Nb from zirconium target for application in immuno-PET. Radiochim Acta 102(5):433–442
Horwitz P, Dietz M, Chiarizia R, Diamond H (1995) Anal Chim Acta 310:63–78
David U, Joel B, Tod W, Eirik K (2003) Rapid sample digestion by fusion and chemical separation of Hf for isotopic analysis by MC-ICPMS. Talanta 59:365
Petrov P, Russell B, Douglas DN, Goenaga-Infante H (2018) Interference-free determination of sub ng kg−1 levels of long-lived 93Zr in the presence of high concentrations (μg kg−1) of 93Mo and 93Nb using ICP-MS/MS. Anal Bioanal Chem 410:1029–1037
Remenec B, Dulanska S, Matel L, Bilohuscin J (2014) Development of a method for the determination of 93Zr and 94Nb in radioactive waste using TEVA_ resin. J Radioanal Nucl Chem 302:117–122
Acknowledgements
The authors would like to thank Prof. Steffen Happel, TRISKEM International for valuable advices, Mihály Veres, ISOTOPTECH Zrt. for ICP-MS measurement possibilities. The work was performed with the professional support of the doctoral student scholarship program of the Co-operative Doctoral Program of the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the KDP-2020 funding scheme. Contract ID: RH/527-14/2021. The research was supported by the European Union and the State of Hungary, co-financed bythe European Regional Development Fund in the project of GINOP_PLUSZ-2.1.1-21-2022-00061.
Funding
Open access funding provided by University of Debrecen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Papp, I., Vajda, N., Bokori, E. et al. Determination of 93Zr, 237Np and Th radionuclides in radioactive waste and mineral samples: extension of the method for determination of actinides. J Radioanal Nucl Chem (2024). https://doi.org/10.1007/s10967-023-09350-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10967-023-09350-0