
Abstract
The green microalga Monoraphidium sp. has potential for biodiesel production since it grows fast and can accumulate high levels of lipids. However, there is a lack of information on the potential use in human nutrition. In this work Monoraphidium sp. was characterized in terms of macronutrients with a special focus on the fatty acid profile of the lipid fraction and on the amino acid profile of the proteins. Furthermore, aiming at nutrient valorization, various methods for lipid extraction that could be used in the production of high quality and safe foods were investigated. To this end, the focus was on cell disruption methods in order to optimize oil recovery. The Monoraphidium sp. biomass had a high protein concentration (44.5 %) and a relatively low lipid concentration (12.5 %) but was rich in ω-3 fatty acids demonstrating its high nutritive value. Regarding cell disruption, ultrasonication and high-speed homogenization were insufficient to disrupt cells under the conditions examined. On the other hand, the effectiveness of the ball milling was regulated via processing time and water-to-biomass ratio and complete cell disruption could be achieved by this method. For lipid extraction, solvent extraction using a mixture of food-grade hexane and ethanol resulted in an oil recovery of 70.4 %, whereas using ethanol alone as an environmentally friendly solvent resulted in an oil recovery equal to 54 %. Supercritical CO2 extraction resulted in a lower oil recovery (25 %), whereas ethanol addition, as a cosolvent to CO2, significantly increased the oil recovery (60 %).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microalgae have been identified as an abundant source of nutrients, including proteins, lipids, vitamins, and minerals (Koyande et al. 2019; Ferreira de Oliveira and Bragotto 2022; Rahman et al. 2022; Sivakumar et al. 2022) and are considered to serve as a sustainable food and feed source. Certain genera of microalgae and cyanobacteria have a high level of proteins, are a valuable source of fatty acids for the human organism, including omega-3 (ω-3) fatty acids, and possess a high level of antioxidants and anti-inflammatory compounds (Xue et al. 2018; Koyande et al. 2019). Other promising characteristics of microalgae are their ability to be cultivated under mild conditions exhibiting though rapid growth rates, their adaptability to field and seasonal conditions with the prospect to be comparable and progressively superior to traditional intensive oil crops in terms of the care they need (Tang et al. 2011; Lin et al. 2019; Ferreira de Oliveira and Bragotto 2022; Hac İsa et al. 2022). Depending on their composition, microalgae may be used in various food applications, including supplements, functional foods, and food ingredients. As a supplement, microalgae can be consumed as a capsule or in powder form to provide a concentrated source of nutrients. Microalgae, can be added to food products to enhance their nutritional value and can also be used as a natural colorant, providing a green hue to food products without the use of synthetic dyes (Dickinson et al. 2017; Koyande et al. 2019; Ferreira de Oliveira and Bragotto 2022).
Microalgal lipids are composed of a variety of fatty acids, including mono- and polyunsaturated fatty acids (PUFA), such as ω-3 and omega-6 (ω-6) fatty acids (Xue et al. 2018; Koyande et al. 2019). Depending on the microalgae species and growth conditions, such as light, temperature, and nutrient levels, the specific composition of microalgal lipids can vary (Xue et al. 2018; Menegazzo et al. 2022). There are numerous potential uses for microalgal lipids, including the production of biofuel, food additives, and personal care products (Dickinson et al. 2017; Koyande et al. 2019; Ferreira de Oliveira and Bragotto 2022). Microalgal lipids can also be used as a source of ω-3 and ω-6 fatty acids in the food industry (Xue et al. 2018; Koyande et al. 2019; Sivakumar et al. 2022).
Proteins are another major nutrient present in microalgae. The content may vary among species and is influenced by environmental conditions and nutrients (Wang et al. 2021). A broad range of protein content has been reported in the literature for microalgae ranging from 6 to 63 % (Chacón-Lee and González-Mariño 2010). When microalgal biomass is considered for human nutrition as a rich protein source, the issue of low digestibility is commonly raised (e.g. Tessier et al. 2021) due to the resistant microalgal cell walls which prevent digestve enzymes for accessing the proteins (Van De Walle et al. 2023).
One of the primary challenges of commercializing microalgae-based products is the efficient extraction of the desired compounds from the cells (Onumaegbu et al. 2018; Zhang et al. 2018; Rahman et al. 2022; Sivakumar et al. 2022). To address this challenge, cell disruption methods have been employed to break down the cell walls and release intracellular contents (Rahman et al. 2022). Such methods include mechanical methods such as bead milling, high-pressure homogenization, ultrasound, and microwave treatment (Oh et al. 2022), chemical methods involving treatment with acids, alkali or heat, osmotic shock, and the use of surfactants (Rahman et al. 2022; Sivakumar et al. 2022) as well as enzymatic methods. Nevertheless, selection of the most appropriate method for cell disruption is difficult because their effectiveness depends greatly on the distinct biology and cell wall characteristics of each microalgal strain (Lee et al. 2017; Oh et al. 2022).
Regarding the lipid extraction processes from microalgae, there are several methods used including solvent, supercritical and enzymatic extraction (Zhou et al. 2022). Traditionally, for solvent lipid extraction polar and non-polar solvents such as hexane, chloroform, butanol, ethanol, methanol, and diethyl ether have been widely employed (Nagappan et al. 2019; Menegazzo et al. 2022; Sivakumar et al. 2022). Supercritical CO2 is a fast, environmentally safe, and often efficient alternative for lipid extraction (Dickinson et al. 2017; Sivakumar et al. 2022) and can be considered a non-toxic green alternative to organic solvents, (Xue et al. 2018; Sivakumar et al. 2022).
Monoraphidium is a freshwater chlorophyte of the family Selenastraceae. The cellular composition of Monoraphidium varies depending on the species and growth conditions such as light intensity (He et al. 2015a, 1983) the green microalga was identified as Monoraphidium sp. The initial ‘natural’ population comprised of singular elongated cells, spindle in shape, straight or curved, sharply pointed at both ends, 12-32 μm long, 2-4 μm wide (Fig. 1a). Water samples collected from the tank were initially inoculated (1 mL) in sterile 50 mL Bold Basal Medium (BBM) (Bold 1949; Bischoff and Bold 1963) and cultivated for 14 days under 16:8 light/dark periods with a light intensity of ~ 50 μmol photons m-2 s-1and temperature of 25 °C. Isolation of unialgal cultures was carried out by serial dilution followed by the micropipette washing technique until unialgal cultures were obtained (Parvin et al. 2007) .

Photomicrographs of (a) the natural Monoraphidium sp. population and (b) Monoraphidium sp. cells cultivated under laboratory conditions. Scale bar: 20 μm
Strain maintenance and lab-scale cultivation conditions (inoculum production)
Under laboratory conditions, Monoraphidium sp. (Fig. 1b) was maintained as unialgal cultures growing in a slightly modified Bold Basal Medium (BBM) (Bold 1949; Bischoff and Bold 1963) at 25 °C and 100 μmol photons m-2 s-1. The light was provided through cool-white LED strips (6400 K, 18 W m-1 and 1700 lumen m-1), with a 16:8-h light:dark photoperiod. The BBM modification involved an increase in sodium nitrate from 250 to 300 mg L-1. To produce sufficient inoculum, Monoraphidium sp. cultures were incubated in shaking incubators (BIOBASE) in 1 and 2 L Erlenmeyer flasks. The cultures were continuous shaken at an agitation rate of 140 rpm and aeration of 0.1 L min-1 with filter-sterilized air (Fig. 2a). After successive cycles of re-culturing, high-density Monoraphidium sp. cultures (~1 g L-1 dry cell mass) were obtained, and 12 L of the culture were condensed through consecutive centrifugation cycles of 5 min at 6000 xg at 15 οC. The resulting pellets were washed twice with sterilized water. Finally, the washed pellets were resuspended in sterilized water at a final volume of 2 L and decanted in an autoclaved sterilized Duran bottle, containing approximately 10 g of dry cell mass (OD@750nm ≈ 10.5).

a) Lab-scale cultivation system, b) semi-pilot scale circular pond and c) pilot scale square-shaped pond
Cultivation of Monoraphidium sp. in a semi-pilot scale circular pond (1st upscaling step)
The initial phase of upscaling for the Monoraphidium sp. culture commenced with a cultivation stage in an open circular pond as illustrated in Fig. 2b. This pond was filled with 100 L fresh UV-sterilised modified Bold's Basal Medium (BBM). The pond received an inoculation of the concentrated inoculum generated within the laboratory-scale units, in accordance with previously outlined procedures. This cultivation was conducted within the greenhouse facilities of the University of Thessaly in Greece, specifically during the summer season of 2021. It commenced with an initial optical density (OD@750nm) of approximately 0.21. The entire culture process spanned a duration of 25 days, encompassing the period from June 18 to July 12. The greenhouse compartment, where the open pond was located, was equipped with a roof vent and with a pad and fan evaporative cooling system aiming to control the greenhouse air temperature levels, while the solar radiation was controlled by a shading screen. The systems were connected to a central greenhouse climate controller (Sercovision, Sercom, The Netherlands) that automatically controlled the above systems in order to obtain air temperature of 25 °C. Furthermore, a pipe network connected to a heat pump was located below each pond in order to automatically maintain the temperature of the pond culture at 25 °C. The autotrophic cultivation of Monoraphidium sp. was achieved by utilizing solar radiation as the illumination source (average daily light integral during the cultivation period: 495 W m-2, average time of photosynthesis per day: 12.69 hours); data of the daily light integral are presented in the Supplementary Material (Fig. S1).
To agitate the culture, a paddle agitator was used at 30 rpm and the temperature was regulated using the greenhouse cooling system in conjunction with aquarium-type heater thermostats placed inside the circular pond. The culture was monitored on a daily basis carrying out OD measurements at 750 and 680 nm, while dry cell weight (DCW) measurements (Psachoulia and Chatzidoukas 2021) were performed every 48 h. The pH of the culture underwent bi-daily assessment and subsequent pH adjustments were executed to achieve a targeted pH level of 7±0.1, employing a 2 M sodium hydroxide (NaOH) solution. Daily inspections of culture purity were conducted by light microscopy. In cases where contaminants, primarily protozoa, were detected, a pH-based treatment approach was adopted. Specifically, the culture pH was significantly reduced to pH 3 (utilizing a 5 M hydrochloric acid (HCl) solution) for a duration of 10 min or elevated to pH 12 (using a 5 M NaOH solution) for an equivalent 10-min interval, following an alternating pattern, as necessitated by the presence of contamination. Subsequently, the pH was readjusted back to the target value of 7. Furthermore, the concentration of nitrate nitrogen (N-NO3) in the medium was periodically measured using the LCK 3339 nitrate cuvette test (HACH) on a UV-vis spectrophotometer (DR 3900, HACH, USA), to ensure nitrogen sufficiency with additional doses of NaNO3 added as required. The BBM was sterilized with a commercial UV sterilizer (P2–110 W UV Sterilizer, Tropical Marine Centre Ltd., UK) for 24 h prior to the inoculation. After 25 days of cultivation the produced microalgae biomass amounted to 1g L-1.
Cultivation of Monoraphidium sp. in a pilot scale square-shaped open pond (2nd upscaling step)
At pilot scale, Monoraphidium sp. was cultivated in a pond system with square dimensions of 3 m per side and a height of 0.5 m, as shown in Fig. 2c. This system was in the same greenhouse as the circular pond employed in the preceding phase. Initially, the pond and the stirring equipment underwent sterilization using a steam vacuum cleaner, generating steam at a temperature of 120°C, to ensure surface cleanliness. Subsequently, the pilot-scale pond was filled with approximately 900 L of freshly UV-sterilized modified BBM. As an additional precautionary measure before inoculation, the pH of the growth medium was temporarily lowered to below pH 3 for one hour. Subsequently, the pH was readjusted to the initial value of the medium using 5M HCl and NaOH solutions, respectively. Subsequently, it was inoculated with the 25-day-old culture obtained from the circular pond that was conveyed via a pum** mechanism (Karcher sp 2 flat pump, 6000 L h-1). After the addition of the inoculum, the depth of the medium was 0.11 m, resulting in a working volume of 1000 L. The functional characteristics of the cultivation system used in this study are described by Bouras et al. (2022). Both the culture monitoring regime as well as the operational strategy followed at this scale unit were the ones employed during the 1st upscaling stage. The pilot-scale autotrophic culture was conducted from July 12 to August 3 (23 days in total). During this timeframe, a total biomass production of 250 g, corresponding to a concentration of 0.25 g L-1, was achieved. The reduced microalgal biomass yield observed during this phase can primarily be attributed to the occurrences of photosaturation and photoinhibition. These phenomena resulted from the intense solar radiation that the Monoraphidium sp. culture experienced during the early stages of pilot cultivation when the culture density was low. Additionally, sporadic episodes of contamination by protozoa and rotifers, although successfully mitigated through pH-based treatments, also contributed to the sustained lower microalgae cell density. The average daily light integral during the cultivation period was 494 W m-2, while the highest light integral, recorded on the 13th day of cultivation was 872 W m-2. Furthermore, the average daily time of photosynthesis was 13.94 hours, and data of the daily light integral are presented in the Supplementary Material (Fig. S2). At the end of the cultivation period the culture was collected through a water pump and transferred to the next harvest stages. The content of microalgae biomass in the culture at harvest was 0.25 % (weight of wet biomass per volume).
Biomass recovery
All biomass analyzed herein or used for further experimentation was obtained by the pilot scale production. Following harvest, a centrifugal separator was used to concentrate the microalgae suspension (Westfalia, Model SA 1-02-175) by 95 % in volume. Using a centrifuge (Sorvall, Model RC-3) with a rotation speed of 4000 rpm for 20 min at 4 °C, the obtained microalgae suspension was further concentrated to a water content of 73–77 % (Fig. 3). The centrifuged wet microalgae biomass was kept in a freezer at −16 oC until the cell disruption tests were performed. It should be noted that the frozen wet biomass was only thawed once for the cell disruption experiments. The Subzero conditions did not affect cell integrity as observed by optical microscopy.

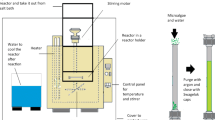
Sequence of treatments and conditions for the biomass recovery, cell disruption and lipids extraction from Monoraphidium sp. (HE-ET: lipid extraction using a mixture of hexane and ethanol, ET: lipid extraction using ethanol, SFE: supercritical CO2 extraction, SFE-ET: supercritical CO2 extraction method using ethanol as a co-solvent)
Cell disruption methods
Microalgae cell disruption examination was carried out using three different methods (a) high-speed homogenization equipped with cell disruption part (Skorupskaite et al. 2017), (b) ultrasound treatment (Duan et al. 2017) and (c) ball milling (Cutshaw et al. 2023):
-
(a)
For the biomass homogenization/disruption an IKA T25 Ultra TURRAX Ultra-homogeniser was employed. For this, 50 g of wet microalgal biomass suspended in 200 mL fresh H2O was treated at a rotor speed of 20000 rpm for 30 min. The sample was sunk in an ice bath in order to prevent overheating that would possibly downgrade the contained macronutrients.
-
(b)
The ultrasound treatments were conducted using a laboratory ultrasonic homogenizer (Hielscher UP-100H, Germany) operated at a power of 100 W and sonication frequency of 30 kHz. The experiment was carried out using 10 g of wet microalgae biomass suspended in 100 mL fresh H2O treated in a sequence of sonication cycles for a total working time of 60 min. The sample container was immersed in an ice bath in order to prevent overheating.
-
(c)
Ball milling experiments were performed with a Fritsch planetary mill (Model pulverisette 5, Fritsch GmbH, Germany) using a grinding bowl of size of 250 mL and 400 g zirconium oxide grinding balls of 0.5 mm diameter and at a 1500 rpm speed. Various grinding times and biomass/water ratios were used as shown in Table 1.
Microalgae cell disruption efficiency was evaluated using an inverted Zeiss LSM 700 confocal microscope (Carl Zeiss, Germany). Both Optical and Confocal Lazer Microscopy were applied (CLSM). For CLSM a minute amount (0.01% w/v) of Nile Red was added to the samples and then the sample was placed on a glass slide which was then covered with a coverslip prior to examination. The magnification used for the observation was ×62. The use of Nile Red allowed for the identification of lipids accumulated in the cellular cytoplasm or over the disrupted cell biomass as well as for the identification of any possible emulsion formation during the cell disruption process.
Lipid extraction
Lipid extraction was performed on cells disrupted by the ball milling process and by applying the conditions BM6 in Table 1. Before extraction the disrupted cell biomass was freeze dried for 48 h using a Christ Gamma 1-20 freeze dryer (Martin Christ Gefriertrocknungsanlagen GmbH) and then ground into a powder within a mortar.
Lipid extraction from ground Monoraphidium sp. was obtained using four different methods: (i) the double solvent extraction method (HE-ET) using a mixture of hexane and ethanol (1:1 ratio) (ii) the single solvent extraction method (ET) using ethanol as a unique solvent (Nagle and Lemke, 1990), (iii) the supercritical CO2 extraction method (SFE), and (iv) the supercritical CO2 extraction method using ethanol as a co-solvent (SFE-ET). The processing steps and experimental conditions for each method are presented in Fig. 3.
The HE-ET method was performed by adding 5 g of freeze dried and crushed microalgal biomass into a conical flask with stopper and 50 mL of hexane-ethanol mixture under stirring for 24 h at 25oC. After the extraction the biomass dispersion in the solvent was centrifuged at 4000 rpm for 15 min (Rotofix 32A, Hettich), the supernatant containing the oil phase was separated, and then the solvents were evaporated in a rotary vacuum evaporator (Buchi R-210 Rotavapor) at 37oC. The ET method was performed similarly to the HE-ET one replacing the mixture of hexane:ethanol with ethanol.
The SFE method was performed in a laboratory scale unit (SFT-110 SFE System – Supercritical Fluid Technologies). For this a sample of 5 g of freeze-dried and crushed microalgal biomass was added into a 20 mL stainless steel extraction vessel. The extraction was performed at 45 oC and 20 MPa. The sample was pressurized with CO2 addition at a flow rate of 10 mL min-1. The chamber was kept pressurized for 30 min and then gradually depressurized within 15 min, while the whole extraction period was 3 h. For the SFE-ET method 5 g of freeze-dried and crushed microalgae biomass were added into a 20 mL stainless steel extraction vessel. The extraction was performed at 20 MPa with CO2 and ethanol (as a co-solvent) addition at a volume ratio of 95 to 5% and flow rate of 10 mL min-1 for 3 h at 45 oC. After the extraction the ethanol from the ethanol–oil mixture was evaporated by a rotary evaporator (R-210 Rotavapor, Buchi) under vacuum at 37 oC. The choice of ethanol as a co-solvent was based on literature data which indicate ethanol as a very effective co-solvent for the supercritical extraction of lipids (Solana et al. 2014). The minimum operating temperature was 45 oC, as the addition of 5% ethanol to CO2 increases the critical temperature of the mixture to 42.5 oC (Adil et al. 2007).
The oil yield of the studied extraction processes was determined using Eq. 1
where, mextract indicates the mass of obtained oil and minitial indicates the weight of the freeze-dried microalgae biomass used for the extraction.
Oil extracted by the BD extraction method was considered as the total oil present in the Monoraphidium sp. biomass.
Additionally, the percent of recovery for each extraction method was evaluated using the ratio of oil yield to oil extracted with BD (Eq. 2)
Biomass composition
The water content of the wet microalgae biomass was determined by drying a 5 g sample in a porcelain crucible in an oven at 105 °C until a constant weight was achieved. The measurement was repeated in triplicate.
The ash content was determined using the samples dried as above. For this the samples were heated at 550 °C for 48 h in a muffle furnace.
The oil content determination of the biomass was performed after cell disruption. This approach was implemented because preliminary experiments demonstrated that without cell disruption only a partial oil recovery was achieved. The total lipid content was determined by the Bligh and Dyer (BD) extraction method using chloroform and methanol as solvents (Bligh and Dyer 1959).
The crude protein content was determined by the Kjeldahl method AOAC 954.01. This method is based on the determination of the total organic nitrogen, and consequently the equivalent protein content, taking into consideration theconversion factor by which the elemental nitrogen is converted to proteins. The conversion factor used was 4.78 based on Lourenco et al., (2004).
The carbohydrates content was determined employing the phenol-sulphuric acid method (AOAC Official Method 988.12 1990).
The fatty acid profile of the biomass lipids, as extracted by the BD method, was determined by gas chromatography. For this a gas chromatography unit (GC Τhermo) equipped with a flame ionization detector (FID) and a DB-FATWAX UI capillary column (30 m x 0.25 mm ID, film thickness 0.25 μm; Agilent J&W Scientific) was used. Peaks were identified by retention times obtained for known standard mixture of 32 fatty acid methyl esters (Supelco). Prior to the analysis the oil was subjected to transesterification to form fatty acid methyl esters (Morrison and Smith 1964).
The amino-acid profile of the freeze-dried microalgal biomass was analyzed using the EZ:faast kit for protein hydrolysates (Phenomenex Inc., USA). Protein hydrolysis was performed using a 6 N HCl solution at 110 °C for 22 h, according to Moore and Stein (1963). Briefly the amino acid analysis procedure consisted of a solid phase extraction step followed by derivatization and liquid/liquid extraction; derivatized samples were then analyzed by gas chromatography with FID detection (Focus GC, Thermo Scientific, USA), equipped with a Zebron ZB-AAA capillary column (10 m x 0.25 mm ID, Phenomenex Inc., USA). Peaks were identified by retention times obtained for known standard amino acid mixture included in the kit.
Results
Biomass composition
Table 2 presents the nutrient composition of Monoraphidium sp. biomass. Under the current culture conditions, the biomass exhibited a total lipid content of 12.5% in dry weight. Proteins and carbohydrates, in addition to lipids, are important intracellular metabolites with their contents being related to each other. In this study the Monoraphidium sp. biomass showed a high protein content (44.5 % dry weight) and a carbohydrate content of 26.6 %, whereas the total mineral (ash) content was 17.4 %.
Fatty acid profile
Table 3 displays the fatty acid profile of the contained lipids. Based on the analysis, the dominant fatty acids detected are palmitic acid (C16:0), oleic acid (C18:1) and alpha-linolenic acid (C18:3 cis 9, 12, 15) with the latter constituting the most abundant fatty acid within the Monoraphidium sp. lipid fraction. Furthermore, the fatty acid profile of Monoraphidium sp. lipid fraction showed a high concentration of unsaturated and ω-3 fatty acids.
Amino acid profile
Table 4 presents the results of the amino acid profile, after GC analysis, of the microalgae protein hydrolysate. It should be noted that the current analysis does not allow the determination of the levels of tryptophane and cysteine. Based on the analysis, 17 amino acids were identified and 8 of them are characterized as essential. In the profile the amino acids with the highest concentration (≥ 10% of total amino acids) were alanine, glycine, leucine, methionine and glutamic acid.
Cell disruption
Figures 4 and 5 illustrate light and CLSM images of Monoraphidium sp. cells following sonication and high-speed homogenization processes. In Fig. 4 it can be observed that ultrasonication, at least under the conditions applied herein, was not able to break effectively all Monoraphidium sp. cells. Regarding the high-speed homogenization Fig. 5 indicated that high-speed homogenization was an insufficient cell disruption method for Monoraphidium sp. cells, as it failed to break any of the cells.

Light (a) and CLSM (b) photomicrographs of Monoraphidium sp. cells after sonication

Light (a) and CLSM (b) photomicrographs of Monoraphidium sp. cells after high-speed homogenization
Figure 6 shows CLSM images of Monoraphidium sp. cells following cell disruption using a planetary ball mill under different conditions. The ball milling method yielded the best results. The efficiency of the process depended strongly on the duration of the treatment as well as on the microalgae biomass/H2O ratio. Figures 6b, c and f show the effect of milling time on cell disruption of wet microalgal biomass with respect to Figure 6a where untreated cells are presented. A short milling time resulted in the disruption of only a few cells (Fig. 6b), but further increase in milling time induced emulsification of the microalgae biomass (Fig 6c and f). Furthermore, the addition of water after milling the wet biomass resulted again in the formation of an emulsion, which induced difficulties in the subsequent oil/water phase separation steps. In the experiments in which water was added to the wet biomass prior to the milling process the breakage of the cells was efficient even after only a few minutes of milling (Figs. 6g and h). With an increase of processing time to 5 min the microalgae cells were completely disrupted (Fig. 6i). These results suggest that a suitable combination of ball-milling parameters can effectively disrupt almost all Monoraphidium sp. cells.

CLSM images of Monoraphidium sp. untreated cells (a) and cells after ball-milling experiments BM1 (b), BM2-a (c), BM2-b (d), BM2-c (e), BM3 (f), BM4 (g), BM5 (j) and BM6 (i)
Oil extraction
The findings regarding oil recovery using the four different extraction methods are presented in Fig. 7. Solvents extraction using food-grade hexane and ethanol (HE-ET) resulted in the highest oil recovery of 70.4 %, even at ambient temperature. Solvent extraction using only ethanol produced a lower recovery of 54 %. However, none of the solvent extraction systems achieved the yields obtained by the BD method (12.5 ± 0.4 %). Regarding oil extraction with supercritical fluids, the SFE process exhibited the lowest oil recovery of 25 % since CO2 extracts only lipids of lower polarity. On the contrary the addition of ethanol as a co-solvent increased the oil recovery value to 60%.

Oil recovery from Monoraphidium sp. extracted by ethanol and hexane (HE-ET), ethanol (ET), supercritical CO2 extraction (SFE) and supercritical CO2 extraction using ethanol as a co-solvent (SFE-ET). Each value is the average of three measurements. The error bars represent the standard deviation
Discussion
Biomass composition
Monoraphidium is a microalgal genus which can accumulate large amounts of lipids and this ability is affected by several factors including nitrogen deprivation (Dhup and Dhawan 2014; Singh et al. 2020; Song et al. 2020), light intensity (He et al. 2015a, 2023).
Regarding the scale-up potential of the lipids production from Monoraphidium sp several aspects have to be considered and improved. First of all, further research is needed for the optimization of cultivation conditions in order to maximize the lipid content of the algal biomass. With respect to the methods employed for cell disruption and lipid extraction, additional research is imperative to implement less energy -intensive and environmentally friendly methods for industrial-scale lipid extraction from Monoraphidium sp. particularly for applications in food and feed industries.
Conclusions
This work extensively studied the potential of utilizing Monoraphidium sp for applications in human nutrition. It was found that Monoraphidium sp. has an interesting nutritional value for human and animal nutrition primarily due to its richness in protein content, methionine, alpha-linolenic acid and ω-3/ω-6 fatty acid ratio. In addition, the lipid fraction of the Monoraphidium sp. studied herein although it is not in a very high concentration, can still be extractable using food grade methods. Aiming to the valorization potential of Monoraphidium sp. for food- grade purposes different cell disruption and lipid extraction methods were explored for the extraction of oil from its biomass. Three mechanical processes were examined to determine their effectiveness in cell disruption. Ultrasonication and high-speed homogenization were inadequate methods to provide a complete disruption of the Monoraphidium sp. cells. However, the use of a ball mill and the appropriate wet biomass/water ratio resulted in complete cell disruption in a relatively short time. For oil extraction, solid-liquid solvent extraction methods using hexane and ethanol mixture or only ethanol, as well as supercritical CO2 extraction using ethanol as a cosolvent, were found to yield high recovery values (54-70 %). Accounting for the scalability of the process, the environmental impact and the initial investment cost, the ET process might be the preferred one when extraction of lipids from dried biomass Monoraphidium sp. is sought at an industrial scale.
However, this study underscores the necessity for further research to increase the lipid content of Monoraphidium sp and to improve lipid recovery through the implementation of more efficient energy-conserving and environmentally sustainable methods at an industrial-scale.
Data availability
Data will be made available on request.
References
Adil IH, Çetin HI, Yener ME, Bayindirli A (2007) Subcritical (carbon dioxide + ethanol) extraction of polyphenols from apple and peach pomaces, and determination of the antioxidant activities of the extracts. J Supercrit Fluids 43:55–63
A.O.A.C. Official Methods of Analysis (1990), 15th edn. Association of Official Analytical Chemists, Washington DC
Bischoff HW, Bold HC (1963) Some soil algae from Enchanted Rock and related algal species. Phycological Studies, University of Texas IV:1-95
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Bold HC (1949) The morphology of Chlamydomonas chlamydogama. Bull Torrey Bot Club 76:101–108
Bouras S, Antoniadis D, Kountrias G, Karapanagiotidis IT, Katsoulas N (2022) Effect of pH on Schizochytrium limacinum production grown using crude glycerol and biogas digestate effluent. Agronomy 12:364
Campanella L, Crescentini G, Avino P (1999) Chemical composition and nutritional evaluation of some natural and commercial food products based on Spirulina. Analusis 27:533–540
Chacón-Lee TL, González-Mariño GE (2010) Microalgae for “healthy” foods-possibilities and challenges. Compr Rev Food Sci Food Saf 9:655–675
Cherian G (2015) Nutrition and metabolism in poultry: role of lipids in early diet. J Anim Sci Biotechnol 6:28
Cutshaw A, Frost H, Uludag-Demirer S, Liu Y, Liao W (2023) Protein extraction, precipitation, and recovery from Chlorella sorokiniana using mechanochemical methods. Energies 16:4809
De Abreu AM, Copetti CLK, Hauschild DB, Di Pietro PF, Wazlawik E (2022) Effects of supplementation with vegetable sources of alpha-linolenic acid (ALA) on inflammatory markers and lipid profile in individuals with chronic kidney disease: a systematic review and meta-analysis. Clin Nutr 41:1434–1444
Delmiro TM, Wilson RRYOV, Melo DMA, Viana ACMG, Mendes BBL, Braga RM (2021) Catalytic flash pyrolysis of Monoraphidium sp. before and after lipid extraction. Algal Res 54:102199
Dhup S, Dhawan V (2014) Effect of nitrogen concentration on lipid productivity and fatty acid composition of Monoraphidium sp. Bioresour Technol 152:572–575
Di Caprio F, Chelucci R, Francolini I, Altimari P, Pagnanelli F (2022) Extraction of microalgal starch and pigments by using different cell disruption methods and aqueous two-phase system. J Chem Technol Biotechnol 97:67–78
Dickinson S, Mientus M, Frey D, Amini-Hajibashi A, Ozturk S, Shaikh F, Sengupta D, El-Halwagi MM (2017) A review of biodiesel production from microalgae. Clean Techn Environ Policy 19:637–668
Dommange X, Tanguy PA, Jolicoeur M (2015) Feasibility of lipid mechanical extraction from viable Monoraphidium minutum. Microalgae Biotech 1:12–19
Duan Z, Tan X, Guo J, Kahehu CW, Yang H, Zheng X, Zhu F (2017) Effects of biological and physical properties of microalgae on disruption induced by a low-frequency ultrasound. J Appl Phycol 29:2937–2946
Dullius A, Fassina P, Giroldi M, Goettert MI, Fernanda C, De Souza V (2020) A biotechnological approach for the production of branched chain amino acid containing bioactive peptides to improve human health: a review. Food Res Int 131:109002
Feng Y, Ma X, Kong B, Chen Q, Liu Q (2023) Ethanol induced changes in structural, morphological, and functional properties of whey proteins isolates: influence of ethanol concentration. Food Hydrocoll 111:106379
Ferreira de Oliveira AP, Bragotto APA (2022) Microalgae-based products: food and public health. Future Foods 6:100157
Furuki T, Maeda S, Imajo S, Hiroi T, Amaya T, Hirokawa T, Ito K, Nozawa H (2003) Rapid and selective extraction of phycocyanin from Spirulina platensis with ultrasonic cell disruption. J Appl Phycol 15:319–324
Garcia-Fernandez JM, Lopez-Ruiz A, Toribio F, Roldan JM, Diez J (1994) Occurrence of only one form of glutamine synthetase in the green alga Monoraphidium braunii. Plant Physiol 104:425–430
Grosso C, Valentão P, Ferreres F, Andrade PB (2015) Alternative and efficient extraction methods for marine-derived compounds. Mar Drugs 13:3182–3230
Hac İsa M, Metin C, Ercan E, Alparslan Y (2022) Effect of different cell disruption methods on lipid yield of Schizochytrium sp. JAOCS 99:129–139
Hammad S, Pu S, Jones PJ (2016) Current evidence supporting the link between dietary fatty acids and cardiovascular disease. Lipids 51:507–517
He Q, Yang H, Hu C (2018) Effects of temperature and its combination with high light intensity on lipid production of Monoraphidium dybowskii Y2 from semi-arid desert areas. Bioresour Technol 265:407–414
He Q, Yang H, Hu C (2016) Culture modes and financial evaluation of two oleaginous microalgae for biodiesel production in desert area with open raceway pond. Bioresour Technol 218:571–579
He Q, Yang H, Wu L, Hu C (2015a) Effect of light intensity on physiological changes, carbon allocation and neutral lipid accumulation in oleaginous microalgae. Bioresour Technol 191:219–228
He Q, Yang H, Xu L, **a L, Hu C (2015b) Sufficient utilization of natural fluctuating light intensity is an effective approach of promoting lipid productivity in oleaginous microalgal cultivation outdoors. Bioresour Technol 180:79–87
Heuzé V., Tran G., Kaushik S. (2020) Soybean meal. In: Feedipedia, a programme by INRAE, CIRAD, AFZ and FAO (2020) 18:25. https://www.feedipedia.org/node/674 (Last updated on March 4, 2020).
Kalogianni EP, Georgiou D, Charisis A, Exarhopoulos E, Tzika P (2023) Valorization of mullet roe by-products for the production of polyunsaturated fatty acids rich oils. JAOCS 100:317–327
Kim KB, Nam YA, Kim HS, Hayes AW, Lee BM (2014) α-Linolenic acid: Nutraceutical, pharmacological and toxicological evaluation. Food Chem Toxicol 70:163–178
Komárek J, Fott B (1983) Chlorophyceae (Grünalgen) Ordnung Chlorococcales. In: Huber-Pestalozzi G (ed) Das phytoplankton des Süβwassers. E. Schweizerbart’sche Verlagsbuchhandlung, Stuttgart, pp 1–1044
Koochi ZH, Jahromi KG, Kavoosi G, Ramezanian A (2023) Fortification of Chlorella vulgaris with citrus peel amino acid for improvement biomass and protein quality. Biotechnol Rep 39:e00806
Koyande AK, Chew KW, Rambabu K, Tao Y, Chu DT, Show PL (2019) Microalgae: a potential alternative to health supplementation for humans. Food Sci Hum Wellness 8:16–24
Lee Y-S, Lee J-Y, Kwon E-B, Kim H-Y, Cho H-J, Kim S-W, Hwang T-K, Byun S-S, Han D-K, Lee J-Y (2009) The effect of human muscle-derived stem cells (MDSC) and glycine-boleucine-lysine-valine-alanine-valine (GIKVAV) on the cryo-injured bladder of nude mouse. Kor J Urol 50:480–485
Lee SY, Cho JM, Chang YK, Oh YK (2017) Cell disruption and lipid extraction for microalgal biorefineries: a review. Bioresour Technol 244:1317–1328
Li D, Zhao Y, Ding W, Zhao P, Xu JW, Li T, Ma H, Yu X (2017) A strategy for promoting lipid production in green microalgae Monoraphidium sp. QLY-1 by combined melatonin and photoinduction. Bioresour Technol 235:104–112
Lin Y, Ge J, Zhang Y, Ling H, Yan X, ** W (2019) Monoraphidium sp. HDMA-20 is a new potential source of α-linolenic acid and eicosatetraenoic acid. Lipids Health Dis 18:56
Lourenço SO, Barbarino E, Lavín PL, Lanfer Marquez UM, Aidar E (2004) Distribution of intracellular nitrogen in marine microalgae: calculation of new nitrogen-to-protein conversion factors. Eur J Phycol 39:17–32
Markou G, Vandamme D, Muylaert K (2014) Microalgal and cyanobacterial cultivation: the supply of nutrients. Water Res 65:186–202
Mattsson L, Sörenson E, Capo E, Farnelid HM, Hirwa M, Olofsson M, Svensson F, Lindehoff E, Legrand K (2021) Functional diversity facilitates stability under environmental changes in an outdoor microalgal cultivation system. Front Bioeng Biotechnol 22:651895
Menegazzo ML, Ulusoy-Erol HB, Hestekin CN, Hestekin JA, Fonseca GG (2022) Evaluation of the yield, productivity, and composition of fatty acids methyl esters (FAME) obtained from the lipidic fractions extracted from Chlorella sorokiniana by using ultrasound and agitation combined with solvents. Biofuels 13:519–526
Moore S, Stein WH (1963) Chromatographic determination of amino acids by the use of automatic recording equipment. Meth Enzymol 6:819–831
Morrison WR, Smith LM (1964) Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J Lipid Res 5:600–608
Nagle N, Lemke P (1990) Production of methyl ester fuel from microalgae Appl Biochem. Biotech 24-25:355–361
Nagappan S, Devendran S, Tsai PC, Dinakaran S, Dahms HU, Ponnusamy VK (2019) Passive cell disruption lipid extraction methods of microalgae for biofuel production – a review. Fuel 252:699–709
Naylor RL, Hardy RW, Buschmann AH, Bush SR, Cao L, Klinger DH, Little DC, Lubchenco J, Shumway SE, Troell M (2021) A 20-year retrospective review of global aquaculture. Nature 591:551–563
Oh YK, Kim S, Ilhamsyah DPA, Lee SG, Kim JR (2022) Cell disruption and lipid extraction from Chlorella species for biorefinery applications: recent advances. Bioresour Technol 366:128183
Onumaegbu C, Mooney J, Alaswad A, Olabi AG (2018) Pre-treatment methods for production of biofuel from microalgae biomass. Renew Sust Energ Rev 93:16–26
Parvin M, Zannat MN, Habib MAB (2007) Two important techniques for isolation of microalgae. Asian Fish Sci 20:117–124
Pineda-Camacho G, de María Guillén-Jiménez F, Pérez-Sánchez A, Raymundo-Núñez LM, Mendoza-Trinidad G (2019) Effect of CO2 on the generation of biomass and lipids by Monoraphidium contortum: a promising microalga for the production of biodiesel. Bioresour Technol Rep 8:100313
Psachoulia P, Chatzidoukas C (2021) Illumination policies for Stichococcus sp. cultures in an optimally operating lab-scale pbr toward the directed photosynthetic production of desired products. Sustainability 13:2489
Qiao T, Zhao Y, Han B, Li T, Zhao P, Xu JW, Huang L, Yu X (2021) Myo-inositol promotes lipid production and nutrients removal by microalga under molasses wastewater. Renew Energy 172:327–335
Rahman M, Hosano N, Hosano H (2022) Recovering microalgal bioresources: a review of cell disruption methods and extraction technologies. Molecules 27:2786
Řezanka T, Nedbalová L, Lukavský J, Střížek A, Sigler K (2017) Pilot cultivation of the green alga Monoraphidium sp. producing a high content of polyunsaturated fatty acids in a low-temperature environment. Algal Res 22:160–165
Safi C, Frances C, Ursu AV, Laroche C, Pouzet C, Vaca-Garcia C, Pontalier PV (2015) Understanding the effect of cell disruption methods on the diffusion of Chlorella vulgaris proteins and pigments in the aqueous phase. Algal Res 8:61–68
Sajjadi B, Raman AAA, Arandiyan H (2016) A comprehensive review on properties of edible and non-edible vegetable oil-based biodiesel: composition, specifications and prediction models. Renew Sust Energ Rev 63:62–92
Sala-Vila A, Fleming J, Kris-Etherton P, Ros E (2022) Impact of α-linolenic acid, the vegetable ω-3 fatty acid, on cardiovascular disease and cognition. Adv Nutr 13:1584–1602
Sargent J, Bell G, Mcevoy L, Tocher D, Estevez A (1999) Recent developments in the essential fatty acid nutrition of fish. Aquaculture 177:191–199
Sheehan J, Dunahay T, Benemann J, Roessler P (1998) A look back at the U.S. Department of Energy’s Aquatic Species Program - Biodiesel from algae. National Renewable Energy Laboratory, Golden, Colorado. Report NREL/TP-580-24190, pp 1-328
Siahbalaei R, Kavoosi G, Noroozi M (2021) Protein nutritional quality, amino acid profile, anti-amylase and anti-glucosidase properties of microalgae: inhibition and mechanisms of action through in vitro and in silico studies. LWT 150:112023
Simopoulos AP (2008) The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med 233:674–688
Singh R, Paliwal C, Nesamma AA, Narula, A, Jutur, P.P. (2020) Nutrient deprivation mobilizes the production of unique tocopherols as a stress-promoting response in a new indigenous isolate Monoraphidium sp. Front Mar Sci 7:575817.
Sivakumar R, Sachin S, Priyadarshini R, Ghosh S (2022) Sustainable production of eicosapentaenoic acid-rich oil from microalgae: towards an algal biorefinery. J Appl Microbiol 132:4170–4185
Skorupskaite V, Makareviciene V, Ubartas M, Karosiene J, Gumbyte M (2017) Green algae Ankistrodesmus fusiformis cell disruption using different modes. Biomass Bioenergy 107:311–316
Solana M, Rizza CS, Bertucco A (2014) Exploiting microalgae as a source of essential fatty acids by supercritical fluid extraction of lipids: comparison between Scenedesmus obliquus, Chlorella protothecoides and Nannochloropsis salina. J Supercrit Fluids 92:311–318
Song X, Zhao Y, Han B, Li T, Zhao P, Xu J-W, Yu X (2020) Strigolactone mediates jasmonic acid-induced lipid production in microalga Monoraphidium sp. QLY-1 under nitrogen deficiency conditions. Bioresour Technol 306:123107
Tang S, Qin C, Wang H, Li S, Tian S (2011) Study on supercritical extraction of lipids and enrichment of DHA from oil-rich microalgae. J Supercrit Fluids 57:44–49
Tessier R, Calvez J, Khodorova N, Gaudichon C (2021) Protein and amino acid digestibility of 15N Spirulina in rats. Eur J Nutr 60:2263–2269
Van De Walle S, Broucke K, Baune MC, Terjung N, Van Royen N, Boukid F (2023) Microalgae protein digestibility: how to crack open the black box? Crit Rev Food Sci Nutr. https://doi.org/10.1080/10408398.2023.2181754
Van Wychen S, Rowland SM, Lesco KC, Shanta PV, Dong T, Laurens LML (2021) Advanced mass balance characterization and fractionation of algal biomass composition. J Appl Phycol 33:2695–2708
Wang Y, Tibbetts SM, McGinn PJ (2021) Microalgae as sources of high-quality protein for human food and protein supplements. Foods 10:3002
WHO/FAO/UNU (World Health Organization) (2007) Protein and amino acid requirements in human nutrition. World Health Organ Tech, Geneva, 180 p
Wilson BA, Pollard RD, Ferguson DS (2014) Nutriential Hazards: Macronutrients: Essential Fatty Acids. In: Encyclopedia of Food Safety, Vo; 3 Elsevier, Amsterdam pp 95–102.
Wu C, **ao Y, Lin W, Li J, Zhang S, Zhu J, Rong J (2017) Aqueous enzymatic process for cell wall degradation and lipid extraction from Nannochloropsis sp. Bioresour Technol 223:312–316
Xue Z, Wan F, Yu W, Liu J, Zhang Z, Kou X (2018) Edible oil production from microalgae: a review. Eur J Lipid Sci Technol 120:1700428
Yu X, Zhao P, He C, Li J, Tang X, Zhou J, Huang Z (2012) Isolation of a novel strain of Monoraphidium sp. and characterization of its potential application as biodiesel feedstock. Bioresour Technol 121:256–262
Zhang Y, Kong X, Wang Z, Sun Υ, Zhu S, Li L, Lv P (2018) Optimization of enzymatic hydrolysis for effective lipid extraction from microalgae Scenedesmus sp. Renew Energy 125:1049–1057
Zhou J, Wang M, Saraiva JA, Martins AP, Pinto CA, Prieto MA, Simal-Gandara J, Cao H, **ao J, Francisco Barba J (2022) Extraction of lipids from microalgae using classical and innovative approaches. Food Chem 384:132236
Funding
Open access funding provided by HEAL-Link Greece. This research was co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH—CREATE—INNOVATE (project code: Τ2ΕΔΚ-02279 – project title: “Human nutrition, animal and fish feeding on microalgae derived products through sustainable photosynthetic autotrophic cultures”.
Author information
Authors and Affiliations
Contributions
Conceptualization: E.P.K., C.C., I.T.K.; methodology: E.P.K., C.C., I.T.K. M.K.; investigation, D.G., S.S., A.C., S.E., G.P. C.S., M.K. G.K. and S.B; resources, E.P.K., C.C., I.T.K. and N.K.; writing—review and editing, E.P.K., C.C., I.T.K., D.G., G.P., M.K.; supervision, E.P.K. project administration, C.C., E. P. K, I.T.K.; funding acquisition, C. C., E.P.K., I.T.K.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 175 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Georgiou, D., Exarhopoulos, S., Charisis, A. et al. Valorization of Monoraphidium sp. microalgal biomass for human nutrition applications. J Appl Phycol 36, 1293–1309 (2024). https://doi.org/10.1007/s10811-024-03191-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-024-03191-4