
Abstract
Background
ESR1 mutations have been identified as mechanism for endocrine resistance and are also associated with a decreased overall survival. We assessed ESR1 mutations in circulating tumor DNA (ctDNA) for impact on outcome to taxane-based chemotherapy in advanced breast cancer patients.
Methods
ESR1 mutations were determined in archived plasma samples from patients treated with paclitaxel and bevacizumab (AT arm, N = 91) in the randomized phase II ATX study. Samples collected at baseline (n = 51) and at cycle 2 (n = 13, C2) were analyzed using a breast cancer next-generation sequencing panel. This study was powered to detect a benefit in progression-free survival (PFS) at six months for patients treated with paclitaxel/bevacizumab compared to historical trials with fulvestrant. PFS, overall survival (OS), and ctDNA dynamics were exploratory analyses.
Results
PFS at six months was 86% (18/21) in patients with an ESR1 mutation detected and 85% (23/27) in wildtype ESR1 patients. In our exploratory analysis, median progression-free survival (PFS) was 8.2 months [95% CI, 7.6–8.8] for ESR1 mutant patients versus 8.7 months [95% confidence interval (CI), 8.3–9.2] for ESR1 wildtype patients [p = 0.47]. The median overall survival (OS) was 20.7 months [95% CI, 6.6–33.7] for ESR1 mutant patients versus 28.1 months [95% confidence interval (CI), 19.3–36.9] for ESR1 wildtype patients [p = 0.27]. Patients with ≥ two ESR1 mutations had a significantly worse OS, but not PFS, compared to those who did not [p = 0.003]. Change in ctDNA level at C2 was not different between ESR1 and other mutations.
Conclusions
Presence of ESR1 mutations in baseline ctDNA might not be associated with inferior PFS and OS in advanced breast cancer patients treated with paclitaxel/bevacizumab.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In metastatic estrogen receptor-positive (ER) breast cancer (MBC), ESR1 mutations have been identified as an important mechanism of resistance to estrogen deprivation therapy by aromatase inhibitors (AIs) and can be detected in approximately 30–50% of MBC patients after prior treatment with AIs in the metastatic setting [16, 20]. Circulating tumor DNA (ctDNA) is now often used as a non-invasive tool for the identification of ESR1 mutations. Additionally, ctDNA is thought to represent the full metastatic landscape which is known to be heterogenic. ctDNA analysis might, therefore, overcome sampling bias introduced by biopsy of one single lesion.
How ESR1 mutations affect sensitivity to subsequent treatment strategies is currently being investigated. Several studies have demonstrated that efficacy of the selective ER down-regulator (SERD) fulvestrant, which is often administered after occurrence of resistance to AIs, is not impaired in patients with ESR1 mutations [6, 19, 21]. More recently, endocrine treatment is being combined with CDK4/6 inhibitors (CDK4/6-i) in the first or second line. Analyses of the PEARL and PALOMA-3 studies have suggested that also CDK4/6-i are effective in patients with ESR1 mutations when combined with fulvestrant [6, 11]. Whether the efficacy of CDK4/6-i when combined with an AI is retained in patients with ESR1 mutations is currently under debate [4, 11]. Also, novel SERDs, which have more favorable pharmacokinetics and ER down-regulating potential than fulvestrant, are being posed as treatment option for patients with ESR1 mutant breast cancer. Elacestrant was recently approved by the FDA for patients an ESR1 mutation who have previously received CDK4/6-I, based on a median PFS improvement of 1.9 months (standard of care endocrine monotherapy) to 3.8 months [3]. In an alternative approach, the PADA-1 trial investigated whether early switching of endocrine therapy based on the occurrence of an ESR1 mutation in plasma, instead of radiological progression, improved outcomes. This study demonstrated that such an approach improves PFS from 5.7 months in patients remaining on AI treatment to 11.9 in patients switching from AI to fulvestrant [2]. However, it is unknown whether this PFS benefit translated into improved OS.
Next to the predictive value of ESR1 mutations for outcome to treatment with AIs, ESR1 mutations have also been associated with worse overall survival [5, 21]. This latter finding suggests that patients with ESR1 mutations have a more aggressive disease biology. The optimal treatment for patients with ESR1 mutations has, however, not been properly determined. For example, it is yet unknown how patients with ESR1 mutations respond to chemotherapy. Therefore, we investigated the outcome to taxane-based chemotherapy in patients with an ESR1 mutation. We analyzed baseline and follow-up plasma samples from the ATX study in which patients were treated with taxane-based chemotherapy after they progressed on endocrine treatment [8].
Methods
Patient population
The ATX study (BOOG-2006-06) was a multicenter, open-label, randomized phase II trial initiated in 2006. Women with confirmed HER2-negative locally recurrent or MBC were eligible. Patients were randomized between paclitaxel (day 1, 8, and 15) plus bevacizumab (days 1 and 15) (AT) at 4-week intervals for six cycles or paclitaxel (day 1 and 8) plus bevacizumab (day 1) plus capecitabine (ATX) at 3-week intervals for eight cycles. For this plasma analysis, we included samples from patients in the AT arm who received prior AI treatment in the adjuvant and/or metastatic setting. We only analyzed samples from the AT arm as the ATX trial showed a difference in progression-free survival (PFS) between the two treatment arms with a median PFS of 11.2 months in the ATX arm and 8.4 months in the AT arm [8]. Moreover, we selected only samples from patients who received an aromatase-inhibitor prior to inclusion. The study was approved by ethical or institutional review boards of the participating hospitals as described previously [8], conducted in agreement with the Declaration of Helsinki and registered with the European Union Drug Regulating Authorities Clinical Trial, number 2006-006058-83, and the Netherlands Trial Register, number NTR1348.
ESR1 mutation analysis
In the trial, plasma was collected from consenting patients at baseline and on day 1 of cycle 2. DNA was isolated from the total amount of plasma that was available, being a median of 1440 uL (range: 60-4000 uL), using the QIAamp Circulating Nucleic Acids kit (Qiagen, Venlo, the Netherlands). Subsequently, the presence of ESR1 mutations was investigated using the Ion Torrent™ Oncomine™ cfDNA Assay for breast cancer, according to protocols and consumables provided by the manufacturer (Life Technologies, Thermo Fisher Scientific, Carlsbad, California, US). The Oncomine™ cfDNA Assay for breast cancer is a targeted panel covering multiple hotspots in 10 genes important in breast cancer (TP53, PIK3CA, ESR1, ERBB2, ERBB3, AKT1, SF3B1, KRAS, EGFR, and FBXW7). For ESR1 the following hotspot mutations are included within this panel: p.D538G, p.Y537S/C/N, p.E380Q, p.V392I, and p.S463P. This NGS panel is equipped with unique molecular identifiers (UMIs) to enable sensitive detection of truly mutated copies. By providing simultaneous information on multiple mutations, the use of a NGS panel enabled us to investigate ESR1 polyclonality and also ESR1 dynamics in plasma. Library preparation for this panel requires 13 uL of DNA input. We used a maximum of 50-ng DNA and if the total DNA yield did not exceed 10 ng, the sample was vacuum dried to a total amount of 13 uL, which was then used for library preparation. Molecular coverage was defined as the count of unique molecules and known hotspot variants were analyzed if they were detected in at least three unique molecules. For follow-up samples, also mutations detected in < 3 molecules were assigned as true variant if the mutation was already identified in a baseline sample. To ensure a sufficient coverage, a total of at least 500 unique molecules had to be sequenced. If this was not reached during the first sequencing run, new libraries were prepared and sequenced and data of the two sequencing runs were summed. This resulted in a theoretical lower limit of detection (LOD) of a variant allele frequency (VAF) of 0.006% (3/500). TP53 mutations were not included in the analysis as we could not exclude the possibility that they had resulted from clonal hematopoiesis [14] and are, therefore, not tumor specific. Germline DNA was not collected within this study. For analysis of ctDNA dynamics, mutations were only included if the molecular coverage at C2 was sufficient to detect the mutation at the baseline frequency.
Assessment of endpoints
The primary endpoint of the ATX study was investigator-assessed PFS, defined as the time from randomization to disease progression or death from any cause, whichever came first. Tumor assessment was performed according to Response Evaluation Criteria in Solid Tumors (RECIST 1.0) every 3 months by computed tomography or magnetic resonance imaging. Secondary endpoints were overall survival (OS), defined as time from randomization to death from any cause, and objective response rate (ORR). ORR was defined as the percentage of complete and partial response confirmed after a minimum of four weeks after first being reported.
Statistical analysis
The primary objective of this study was to investigate whether patients with an ESR1 mutation benefit from taxane-based chemotherapy. Therefore, we hypothesized that taxane-based treatment would increase the PFS at 6 months when compared to historical trials with fulvestrant, the current standard of treatment in patients with an ESR1 mutation. The PFS percentage at 6 months after start of treatment with fulvestrant in this specific patient category is approximately 40% [6]. For this study, we proposed that a PFS at 6 months of approximately 75% would justify further testing of chemotherapy versus fulvestrant in a phase III randomized trial [12]. To test this hypothesis, we used a Simon two-stage design with p0 = 0.4 and p1 = 0.75 with a power of 0.8 and a type 1 error rate of 0.05. As such, at least 8 out of 14 patients with an ESR1 mutation had to be free of progression at 6 months to conclude that chemotherapy is sufficiently promising in this patient group to warrant further investigation. Using an exploratory analysis, we compared PFS and OS in patients with and without a detectable ESR1 mutation. Descriptive statistics were calculated for variables of interest. Wilcoxon signed-rank test was performed for univariate analyses of continuous variables and a Fisher exact test or chi-square test was used for categorical variables. The duration of time to event was estimated using the Kaplan–Meier method. Statistical tests were two-sided and considered statistically significant when p < 0.05. IBM SPSS STATISTICS 25 (ICM Corp, Armonk, NY) was used for survival analysis and descriptive statistics. Prism™ software (GraphPad Software, La Jola, Ca) was used for the statistical analyses of ctDNA dynamics.
Results
ESR1 mutation analysis
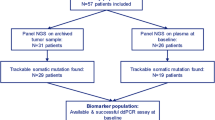
In the ATX trial, 156 patients were randomized to paclitaxel/bevacizumab (AT arm), of whom 85% were ER-positive. Of those, 91 patients received prior treatment with an AI in the adjuvant and/or metastatic setting. Residual baseline plasma samples were available of 51 patients (56%), since plasma had already been used for other purposes [9, 10]. Samples were subjected to NGS. Three samples did not reach a total molecular coverage of ≥ 500 molecules, despite duplicate library preparation and sequencing and were, therefore, excluded from the analysis (Fig. 1). In total 48 patients were included, of which 21 (44%) had a detectable ESR1 mutation. Adjuvant endocrine treatment was not different among patients with ESR1 mutant and wildtype tumors (p = 0.28), whereas patients with an ESR1 mutation tended to have received palliative endocrine treatment more often 86% vs 67% (p = 0.19). The median cfDNA concentration was 42-ng/mL plasma (range: 10–125 ng/mL) among patients with ESR1 mutant tumors and 24-ng/mL plasma (range: 6–133 ng/mL) among patients with ESR1 wildtype tumors (p = 0.1). Of the patients with a detectable ESR1 mutation, eight patients (38%) had multiple ESR1 mutations (Fig. 2).

CONSORT diagram demonstrating samples analyzed in the ATX trial

ESR1 variants in patients with detectable ESR1 mutations
ESR1 mutations and PFS
One patient died before progression and three patients started other systemic therapy before radiological progression. PFS at 6 months was 86% (18/21) in patients with an ESR1 mutation detected and 85% (23/27) in wildtype ESR1 patients. In patients with a detectable ESR1 mutation, median PFS was 8.2 months (95% CI 7.6–8.8 months) and in wildtype ESR1 patients, median PFS was 8.7 months (95% CI 8.3–9.2 months, log rank p = 0.47, Fig. 3A). When split by the median ESR1 VAF, PFS was not different between patients with a ESR1 VAF above the median (median 8.1 months, 95% CI 7.8–8.5 months) and a ESR1 VAF below the median (median 8.4 months, 95% CI 7.2–9.6 months, log rank p = 0.581). Also, PFS was not significantly different among patients with multiple ESR1 mutations (median: 7.5 months, 95% CI 5.2–9.8 months) versus patients without or one ESR1 mutation(s) (median 8.6 months, 95% CI 8.2–9.1 months, log rank p = 0.35). We did not analyze a possible relation of the presence of specific ESR1 variants with outcome as the numbers were too low Table 1.

A Progression-free survival in patients treated with paclitaxel/bevacizumab by ESR1 mutation status. B Overall survival in these patients by ESR1 mutation status
ESR1 mutations, OS, and objective response rate
We subsequently investigated the association between ESR1 mutation status and OS, with a total of 42 deaths. In patients with a detectable ESR1 mutation, median OS was 20.7 months (95% CI 6.6–33.7 months), whereas in wildtype ESR1 patients, median OS was 28.1 months (95% CI 19.3–36.9 months, log rank p = 0.27, Fig. 3B). When split by the median ESR1 VAF, OS was not different between patients with a high ESR1 VAF (median 20.7 months, 95% CI 9.1–32.3 months) and a low ESR1 VAF (median 17.4 months, 95% CI 6.9–30.9 months, log rank p = 0.7). Interestingly, OS was significantly shorter in patients with multiple ESR1 mutations (median: 14.6 months, 95% CI 8.1–21.0 months) when compared to those without or with one ESR1 mutation (median 28.9 months, 95% CI 20.4–37.5 months, p = 0.003, Figure S1). Moreover, the total number of mutations in any of the ten genes analyzed was significantly correlated with overall survival in univariate cox proportional hazard regression analysis (HR 1.26, 95% CI 1.04–1.54, p = 0.017). In the ESR1 wildtype group, one patient (4%) had a complete response to treatment, whereas none of the patients in the ESR1 mutant group had a complete response. Overall, the ORR was lower in patients with an ESR1 mutation (40%) than in wildtype ESR1 patients (65%), but this was not significant (p = 0.136, Table 2).
ctDNA dynamics
Of the 21 patients with an ESR1 mutation, follow-up samples were available from 13 patients (62%). Of those, 11 patients also had a PIK3CA or AKT1 mutation. At C2, ESR1 mutations were undetectable in four patients (31%). In total, 24 ESR1 and 18 PIK3CA or AKT1 mutations were identified in baseline samples and assessed in follow-up samples. Those PIK3CA and AKT1 mutations were considered as a surrogate marker for ctDNA load and used to calculate whether ESR1 mutant subclones showed a differential response to treatment. Of the 24 individual ESR1 mutations, 16 (67%) were not detected in follow-up samples. Additionally, of the 20 PIK3CA or AKT1 mutations, 9 (45%) were not detected in follow-up samples (Fig. 2A). We defined the circulating DNA ratio (CDR) as the ratio of mutation abundance on treatment (C2) relative to baseline, in which CDR represents the ratio of ctDNA levels at C2 to ctDNA levels at baseline. All patients had a CDR < 1 indicating a fall in ctDNA. The CDR was not different between ESR1 mutations and PIK3CA or AKT1 mutations (p = 0.547, Fig. 2B), suggesting there was no differential response to treatment in ESR1 mutant subclones versus PIK3CA or AKT1 mutant subclones. The results were similar for mutant copies/mL plasma (Figure S2). Because of the low patient number from whom follow-up samples were available, analysis of CDR and outcome to paclitaxel/bevacizumab was not performed as shown in Fig. 4.

A Dynamics of the VAF of ESR1 and PIK3CA or AKT1 mutations between baseline and cycle 2, day 1, and differences between the VAF were calculated using the Wilcoxon signed-rank test. Connecting lines are colored per patient. B CDR of the VAF from ESR1 mutation vs PIK3CA or AKT1 mutations. Differences between the CDR were calculated using the Wilcoxon signed-rank test. Line at median
Discussion
To investigate the clinical impact of ESR1 mutations in cfDNA on the efficacy of chemotherapy in metastatic ER+/HER2- breast cancer patients, we analyzed plasma samples from advanced ER+ breast cancer patients that were treated with first-line paclitaxel/bevacizumab in the ATX trial. We demonstrated that paclitaxel/bevacizumab might be an effective treatment strategy in patients independent of ESR1 mutations status. Based on comparison with literature, which suggests the PFS at 6 months after treatment with fulvestrant is 40% [6], treatment with paclitaxel/bevacizumab has a superior PFS at six months of 85% regardless ESR1 mutation status. The results of this retrospective, exploratory study suggest that the efficacy of chemotherapy is retained in patients with one or more ESR1 mutations, although our study was not powered to detect a difference in survival between patients with ESR1 mutations and wildtype ESR1 patients.
We observed that chemotherapy resulted in a decline of ESR1 mutant ctDNA after one cycle of chemotherapy. For the two patients with confirmed progression at 6 months after treatment initiation, we did not have a C2 sample available to investigate whether ctDNA dynamics could have predicted this outcome. In the majority of ESR1 mutant patients, the mutation could not be detected after one cycle of treatment. Although we used plasma samples containing an unknown amount of wildtype contamination, we observed a fall in ctDNA levels of both ESR1 and PIK3CA or AKT1 mutations after 1 cycle of treatment, suggesting that this fall in ctDNA load represents a decreased tumor load rather than pre-analytical variation. Additionally, we found that there was no difference in suppression of ESR1 mutations, which were often subclonal, when compared to PIK3CA or AKT1 mutations, suggesting the anti-proliferative effect of chemotherapy does not differentially affect cells that have acquired an ESR1 mutation.
We found that the presence of multiple ESR1 mutations, but not single ESR1 mutations, was associated with an impaired OS, although in a small set of patients. ESR1 mutations were previously identified as prognostic biomarker in the BOLERO-2 study [5], as well as in the EFECT and SoFEA trial [21]. The prognostic value of having polyclonal ESR1 mutations was also observed in the BOLERO-2 trial [5]. Patients with polyclonal ESR1 mutations in ctDNA might have tumors that are heterogeneous and, therefore, more likely to consist of other resistant subclones besides ESR1. To substantiate this finding, we took the total number of mutations in any of the ten genes that were analyzed as a surrogate for heterogeneity and demonstrated that this total number of mutations was significantly correlated with overall survival. This finding highlights the importance of understanding tumor heterogeneity in a non-invasive manner and emphasize approaches to target or even to prevent this heterogeneity.
Previous studies have suggested that ESR1 variants differentially impact sensitivity to treatment. In analyses from the PALOMA-3 and SoFEA study, patients harboring the ESR1 p.Y537S had the shortest PFS on fulvestrant and this variant was enriched after treatment with fulvestrant with or without CDK4/6-i (16). Also in patients treated with everolimus and exemestane, ESR1 p.Y537S tended to be negatively associated with PFS and OS [7]. As our study was limited by the sample size, we did not perform subgroup analysis for specific ESR1 variants. In general, the ESR1 variant with the highest VAF was the mutation that remained detectable in patients with polyclonal ESR1 mutations, suggesting the decline in VAF is a reflection of decreasing tumor load rather than a differential response.
As a hypothesis, detection of ESR1 mutations might be useful to determine whether sensitivity to endocrine treatment might be restored after anti-proliferative agents such as CDK4/6-i or chemotherapy. The CHRONOS study has demonstrated that patients with metastatic colorectal cancer [17], who have progressed due to RAS/RAF mutations after previous response to anti-EGFR treatment and who lacked detectable BRAF or RAS mutations in ctDNA after progression to the last anti-EGFR-free regimen, could be successfully re-challenged with anti-EGFR treatment. As such, it might be investigated whether endocrine-resistant advanced breast cancer patients in whom ESR1 mutations become undetectable after chemotherapy can be successfully re-introduced to endocrine treatment.
This retrospective study contains several flaws. The analysis of the impact of the ESR1 mutational status on outcome of chemotherapy was limited due to the relatively low sample size at baseline and the number of patients with samples available at C2. The small sample size also hampered us from conducting various subanalyses investigating the differential impact of ESR1 variants or variants in other genes on outcome. Also, we were not able to investigate the effect of ctDNA dynamics on outcome. As such, our findings are exploratory and should be interpreted with caution. The lack of plasma samples at progression precluded the question, whether chemotherapy can lead to definite clearance of ESR1 mutations enabling re-challenge with endocrine treatment. Furthermore, bevacizumab is no longer added to taxane-based chemotherapy in current practice. The PFS benefit of adding bevacizumab to taxane-based treatment [12] could not be validated in later studies and did not translate into OS benefit [13, 15] resulting in the FDAs withdrawal of the breast cancer indication for bevacizumab. Although we could not propose a mechanism by which VEGF inhibition could have affected the cytostatic effect of paclitaxel on ESR1 mutant cells, the effect of this limitation on the results of this study remain unknown. The limited sample size of this study might also be an explanation for our finding that there was no statistical difference in receipt of prior endocrine treatment between ESR1 mutant and wildtype patients, while literature indicates that ESR1 mutations mainly occur after treatment with an AI in the metastatic setting [1, 16, 18, 20]. In conclusion, we found no association between the presence of ESR1 mutations in baseline ctDNA and PFS in patients treated with paclitaxel/bevacizumab. Therefore, the results of this retrospective, exploratory study suggest that the efficacy of chemotherapy is retained in patients with one or more ESR1 mutations. We further found that having polyclonal disease was associated with impaired OS. Lastly, we found that in some patients, ESR1 mutations become undetectable after one cycle of chemotherapy. Whether this results in restoration of endocrine sensitivity remains to be investigated.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, Nik-Zainal S, Steenbruggen TG, Tjan-Heijnen VCG, Labots M, van Riel JMGH, Bloemendal HJ, Steeghs N, Lolkema MP, Voest EE, van de Werken HJG, Jager A, Cuppen E, Sleijfer S, Martens JWM (2019) The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 51:1450–1458. https://doi.org/10.1038/s41588-019-0507-7
Bidard F-C, Hardy-Bessard A-C, Dalenc F, Bachelot T, Pierga J-Y, de la Motte RT, Sabatier R, Dubot C, Frenel J-S, Ferrero JM, Ladoire S, Levy C, Mouret-Reynier M-A, Lortholary A, Grenier J, Chakiba C, Stefani L, Plaza JE, Clatot F, Teixeira L, D’Hondt V, Vegas H, Derbel O, Garnier-Tixidre C, Canon J-L, Pistilli B, André F, Arnould L, Pradines A, Bièche I, Callens C, Lemonnier J, Berger F, Delaloge S, Bidard F-C, Pistilli B, Dalenc F, Bachelot T, De La Motte RT, Sabatier R, Dubot C, Frenel J-S, Ferrero J-M, Ladoire S, Levy C, Mouret-Reynier M-A, Hardy-Bessard A-C, Lortholary A, Grenier J, Chakiba C, Stefani L, Soulie P, Jacquin J-P, Plaza JE, Clatot F, Teixeira L, D’Hondt V, Vegas H, Derbel O, Garnier Tixidre C, Delbaldo C, Moreau L, Cheneau C, Paitel J-F, Bernard-Marty C, Spaeth D, Genet D, Moullet I, Bonichon-Lamichhane N, Deiana L, Greilsamer C, Venat-Bouvet L, Delecroix V, Melis A, Orfeuvre H, Nguyen S, Legouffe E, Zannetti A, Le Scodan R, Dohollou N, Dalivoust P, Arsene O, Marques N, Petit T, Mollon D, Dauba J, Bonnin N, Morvan F, Gardner M, Marti A, Levache C-B, Lachaier E, Achille M, Valmar C, Bouaita R, Medioni J, Foa C, Bernard-Marty C, Del Piano F, Gozy M et al (2022) Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol 23:1367–1377. https://doi.org/10.1016/s1470-2045(22)00555-1
Bidard F-C, Kaklamani VG, Neven P, Streich G, Montero AJ, Forget F, Mouret-Reynier M-A, Sohn JH, Taylor D, Harnden KK, Khong H, Kocsis J, Dalenc F, Dillon PM, Babu S, Waters S, Deleu I, García Sáenz JA, Bria E, Cazzaniga M, Lu J, Aftimos P, Cortés J, Liu S, Tonini G, Laurent D, Habboubi N, Conlan MG, Bardia A (2022) Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, human epidermal growth factor receptor 2–negative advanced breast cancer: results from the randomized phase III EMERALD trial. J Clin Oncol 40:3246–3256. https://doi.org/10.1200/jco.22.00338
Bidard FC, Hardy-Bessard A-C, Bachelot T (2021) Fulvestrant-palbociclib vs continuing AI-palbociclib upon detection of circulating ESR1 mutation in HR+ HER2- mBC patients: results of PADA-1, a UCBG-GINECO randomized phase 3 trial. In: San Antonio Breast Cancer Symposium. Virtual
Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, Bhatt T, Patel P, Voi M, Gnant M, Hortobagyi G, Baselga J, Moynahan ME (2016) Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2:1310–1315. https://doi.org/10.1001/jamaoncol.2016.1279
Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S, Loibl S, Jiang J, Bartlett CH, Koehler M, Dowsett M, Bliss JM, Johnston SRD, Turner NC (2016) Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 34:2961–2968. https://doi.org/10.1200/jco.2016.67.3061
Kruger DT, Jansen MPHM, Konings IRHM, Dercksen WM, Jager A, Oulad Hadj J, Sleijfer S, Martens JWM, Boven E (2020) High ctDNA molecule numbers relate with poor outcome in advanced ER+, HER2− postmenopausal breast cancer patients treated with everolimus and exemestane. Mol Oncol 14:490–503. https://doi.org/10.1002/1878-0261.12617
Lam SW, de Groot SM, Honkoop AH, Jager A, ten Tije AJ, Bos MM, Linn SC, van den Bosch J, Kroep JR, Braun JJ, van Tinteren H, Boven E, Dutch Breast Cancer Research G (2014) Paclitaxel and bevacizumab with or without capecitabine as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a multicentre, open-label, randomised phase 2 trial. Eur J Cancer 50:3077–3088. https://doi.org/10.1016/j.ejca.2014.10.008
Lam SW, Nota NM, Jager A, Bos MMEM, van den Bosch J, van der Velden AMT, Portielje JEA, Honkoop AH, van Tinteren H, Boven E, the ATXtt (2016) Angiogenesis- and hypoxia-associated proteins as early indicators of the outcome in patients with metastatic breast cancer given first-line bevacizumab-based therapy. Clin Cancer Res 22:1611–1620. https://doi.org/10.1158/1078-0432.Ccr-15-1005
Lam SW, van der Noort V, van der Straaten T, Honkoop AH, Peters GJ, Guchelaar H-J, Boven E (2018) Single-nucleotide polymorphisms in the genes of CES2, CDA and enzymatic activity of CDA for prediction of the efficacy of capecitabine-containing chemotherapy in patients with metastatic breast cancer. Pharmacol Res 128:122–129. https://doi.org/10.1016/j.phrs.2017.08.005
Martin M, Zielinski C, Ruiz-Borrego M, Carrasco E, Turner N, Ciruelos EM, Muñoz M, Bermejo B, Margeli M, Anton A, Kahan Z, Csöszi T, Casas MI, Murillo L, Morales S, Alba E, Gal-Yam E, Guerrero-Zotano A, Calvo L, de la Haba-Rodriguez J, Ramos M, Alvarez I, Garcia-Palomo A, Huang Bartlett C, Koehler M, Caballero R, Corsaro M, Huang X, Garcia-Sáenz JA, Chacón JI, Swift C, Thallinger C, Gil-Gil M (2021) Palbociclib in combination with endocrine therapy versus capecitabine in hormonal receptor-positive, human epidermal growth factor 2-negative, aromatase inhibitor-resistant metastatic breast cancer: a phase III randomised controlled trial—PEARL☆. Ann Oncol 32:488–499. https://doi.org/10.1016/j.annonc.2020.12.013
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357:2666–2676. https://doi.org/10.1056/NEJMoa072113
Pivot X, Schneeweiss A, Verma S, Thomssen C, Passos-Coelho JL, Benedetti G, Ciruelos E, von Moos R, Chang H-T, Duenne A-A, Miles DW (2011) Efficacy and safety of bevacizumab in combination with docetaxel for the first-line treatment of elderly patients with locally recurrent or metastatic breast cancer: results from AVADO. Eur J Cancer 47:2387–2395. https://doi.org/10.1016/j.ejca.2011.06.018
Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, Abida W, Juluru K, De Bruijn I, Hou C, Venn O, Lim R, Anand A, Maddala T, Gnerre S, Vijaya Satya R, Liu Q, Shen L, Eattock N, Yue J, Blocker AW, Lee M, Sehnert A, Xu H, Hall MP, Santiago-Zayas A, Novotny WF, Isbell JM, Rusch VW, Plitas G, Heerdt AS, Ladanyi M, Hyman DM, Jones DR, Morrow M, Riely GJ, Scher HI, Rudin CM, Robson ME, Diaz LA, Solit DB, Aravanis AM, Reis-Filho JS (2019) High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 25:1928–1937. https://doi.org/10.1038/s41591-019-0652-7
Robert NJ, Diéras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, Perez EA, Yardley DA, Chan SYT, Zhou X, Phan S-C, O’Shaughnessy J (2011) RIBBON-1: randomized, double-blind, placebo-controlled, phase iii trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2–negative, locally recurrent or metastatic breast cancer. J Clin Oncol 29:1252–1260. https://doi.org/10.1200/jco.2010.28.0982
Robinson DR, Wu Y-M, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM (2013) Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45:1446–1451. https://doi.org/10.1038/ng.2823
Sartore-Bianchi A, Pietrantonio F, Lonardi S, Mussolin B, Rua F, Fenocchio E, Amatu A, Corallo S, Manai C, Tosi F, Manca P, Daniel F, Torri V, Vanzulli A, Cappello G, Marchiò C, Sapino A, Marsoni S, Siena S, Bardelli A (2021) Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: the CHRONOS trial. J Clin Oncol 39:3506–3506. https://doi.org/10.1200/JCO.2021.39.15_suppl.3506
Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, Fenwick K, Kozarewa I, Lopez-Knowles E, Ribas R, Nerurkar A, Osin P, Chandarlapaty S, Martin L-A, Dowsett M, Smith IE, Turner NC (2015) Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Trans Med 7:313ra182-313ra182. https://doi.org/10.1126/scitranslmed.aac7551
Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, Aimi J, Derynck MK, Chen M, Chan IT, Amler LC, Hampton GM, Johnston S, Krop I, Schmid P, Lackner MR (2016) Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 7:11579. https://doi.org/10.1038/ncomms11579
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S (2013) ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45:1439–1445. https://doi.org/10.1038/ng.2822
Turner NC, Swift C, Kilburn L, Fribbens C, Beaney M, Garcia-Murillas I, Budzar AU, Robertson JFR, Gradishar W, Piccart M, Schiavon G, Bliss JM, Dowsett M, Johnston SRD, Chia SK (2020) ESR1 mutations and overall survival on fulvestrant versus exemestane in advanced hormone receptor-positive breast cancer: a combined analysis of the phase III SoFEA and EFECT trials. Clin Cancer Res 26:5172. https://doi.org/10.1158/1078-0432.Ccr-20-0224
Acknowledgements
None
Funding
The ctDNA analysis of this study were funded by the Dutch Cancer Society (no. NKB-EMCR-2016-108154). The ATX trial was funded by F. Hofmann-la Roche Ltd (Woerden, the Netherlands).
Author information
Authors and Affiliations
Contributions
SWL, AJ, and EB were involved in the execution of the ATX trial and collection of clinical data. MKB, GM, JCH, CMB, and EdJ were responsible for the cell-free DNA analyses. All authors were involved in data interpretation. MKB wrote the first draft of the manuscript which was reviewed and edited by all authors. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interest
M.K. Bos received research funding from Dutch Cancer Society (no. NKB-EMCR-2016–108154), S.W. Lam: none, G. Motta: none, J.C.A. Helmijr: none, C.M. Beaufort: none, E. de Jonge: none, J.W.M. Martens received sponsorship for research from Pfizer, Menarini, GSK Cytotrack, Cergentis, payment for consultancy from Novartis, and payment for presentation from Roche, E. Boven: none, M.P.H.M. Jansen: none, A. Jager: none, and S. Sleijfer: none.
Ethical approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of all participating centers. Written informed consent was obtained before study enrollment. This trial is registered with the European Union Drug Regulating Authorities Clinical Trials, number 2006–006058-83, and the Netherlands Trial Register, number NTR1348.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bos, M.K., Lam, S.W., Motta, G. et al. Plasma ESR1 mutations and outcome to first-line paclitaxel and bevacizumab in patients with advanced ER-positive/HER2-negative breast cancer. Breast Cancer Res Treat 200, 271–279 (2023). https://doi.org/10.1007/s10549-023-06965-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-023-06965-5