
Abstract
Applying the most appropriate sampling method is essential for estimating population size. Sampling methods and techniques to estimate abundance may be limited by environmental characteristics, species traits, specific requirements of the techniques, or the economic resources to carry out the sampling. Thus, evaluating multiple sampling methods in monitoring populations is essential for establishing effective conservation strategies. In this study, we compare two of the most commonly used sampling methods with the red fox (Vulpes vulpes) as the type species. On the one hand, we compared the minimum number of individuals (NI) detected by camera trap**, identifying individuals by morphological characteristics with the minimum number of individuals detected by DNA faeces and a set of 16 microsatellites. On the other hand, we estimated abundance by performing an N-mixture model using information from camera-traps to study the relationship between abundance and the minimum number of individuals detected. Results showed that the minimum NI provided by camera trap** was slightly higher than that of DNA faecal genoty**, with 23.66 and 19 individuals, respectively. In addition, abundance and NI detected by camera trap** showed a positive relationship. In contrast, there was a non-significant negative relationship between NI detected by faecal DNA and abundance estimates. Our results suggest using the minimum number of photo-identified individuals as a reliable index to study variation in red fox abundance when other advanced methods cannot be implemented in the study of population size. However, it is necessary to improve the methods of faecal sampling to study the relationship with camera-trap data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent acceleration of biodiversity loss urgently requires the development of monitoring programmes to understand trends and spatial patterns of the abundance of wildlife species. Among these, the order Carnivora is among the most threatened groups worldwide (Gittleman et al. 2001). Carnivores are emblematic species with an important ecological role; however, they also cause conservation conflicts with human and their activities (Linnell and Strand 2000; Prugh et al. 2009; Ritchie et al. 2012). Many carnivore species are elusive and solitary mesopredators (hereafter mesocarnivores), with nocturnal habits, large territories and low population densities. These characteristics make non-invasive survey methods highly suitable for studying and monitoring these species. Camera trap** and DNA-genoty** are currently the most relevant non-invasive sampling methods applied to monitor mesocarnivores and analyse their abundance assisted by different analytical techniques (Srbek-Araujo and Chiarello 2005; Trolle et al. 2007; O’Connell et al. 2011; Sollmann et al. 2013; Rodgers et al. 2014). However, these methods are unreliable in all scenarios because environmental characteristics and species traits may limit them, as well as the specific requirements of the techniques to estimate species abundance or the economic resources available to carry out the sampling. Therefore, because different sampling methods and techniques can be applied to estimate mesocarnivore abundance in different regions or seasons, it is necessary to assess the relationship between these methods and how they can be selected in terms of their sampling effort, economic costs, precision and accuracy of abundance estimates (Silveira et al. 2003; Gaidet-Drapier et al. 2006; Gompper et al. 2006; Balme et al. 2009).
Abundance can be assessed using population size estimates or indices of relative abundance. Among the former, capture-recapture methods are widely used by biologists to estimate parameters of wildlife population (Pollock 1976) using the capture histories of identified individuals to draw the detection probability. Camera trap** and non-invasive DNA sampling are among the most widespread techniques used to identify individuals (Karanth and Nichols 1998; Jackson et al. 2006; Mondol et al. 2009; Galaverni et al. 2012). However, other techniques such as N-mixture methods can be used when individuals cannot be individually identified. N-mixture models use data from spatially replicated count surveys to effectively estimate population sizes while accounting for the detection process (Royle 2004; Ficetola et al. 2018; Kidwai et al. 2019; Costa et al. 2020). However, these methods imply a great effort and resources as they require a high number of spatial replicates to account for imperfect detection (instance >20 sites; Kéry and Schaub 2012) and a high probability of detection (Royle 2004; Veech et al. 2016) to obtain reliable abundance estimates. Under these limitations for estimating abundance, relative abundance indices are helpful and convenient for studying population size. Indices of relative abundance are positively correlated with the population size of the species (Caughley and Sinclair 1994), so changes in index values reflect a change in actual abundance (Romesburg 1981; Anderson 2003). Among relative abundance indices faeces counts along transects and capture rates by camera-traps are the most important. However, using relative abundance indices should be taken with caution because the monotonic relationship with true abundance may not be constant and positive over time and across habitats. In addition, different indices may show different sensitivities to the same habitat characteristics; therefore, relative abundance estimates will depend on the index used. In addition, the indices do not control the probability of detection of a species as do the technique mentioned above (CR and N-mixture methods), which may influence abundance estimates. Moreover, we should consider that the efficient use of camera trap** and DNA faeces sampling might be limited under some circumstances that could affect abundance estimation. For example, environments with high humidity and temperature poorly preserve DNA in faeces (Murphy et al. 2007). Temperature differences can also lead to reduced detection of animals and undesirable detection biases in camera-traps triggered by ‘heat-in-motion’ (Meek et al. 2015).
Identifying relative abundance indices that follow a constant relationship with abundance without depending on the sampling method used, habitat characteristics or probability of detection is of great interest when other advanced methods cannot be implemented in the study of population size. Here, we compare the relationship between camera trap** and DNA faecal sampling to study population size during the same spatio-temporal settings. We compared the minimum number of individuals (NI) identified by each sampling method in several areas covering different habitat characteristics. We propose the minimum NI as a relative index of abundance and study the relationship of this index with abundance estimation. In particular, we used N-mixture models (Royle 2004) to estimate abundance using camera-traps. Previous studies focused on carnivore species with spotted fur or animals marked to facilitate the identification of individuals using camera-traps (Karanth and Nichols 1998; Silveira et al. 2003; Soisalo and Cavalcanti 2006; Mosquera et al. 2016). We used one of the most abundant mesocarnivore species in Europe and particularly in the Iberian Peninsula (Lloyd 1980), the red fox (Vulpes vulpes). Although the red fox lacks a distinct fur pattern, all individuals show distinctive characteristics (i.e. body and tail coat; muzzle, head and ears shape; leg and paw marking; injuries) that allow individual for identification (Sarmento et al. 2009; Dorning and Harris 2019). Furthermore, territory marking by foxes’ character facilitates faeces sampling (Goszczyński 1990). These characteristics make the fox a model species to test non-invasive sampling techniques in carnivores. The abundance of red foxes is not well-documented in all areas of the Iberian Peninsula. However, some studies have tested different non-invasive sampling techniques to determine its abundance (Jiménez et al. 2017; Jiménez et al. 2019a; Jiménez et al. 2019b). The red fox has an critical ecological role in seed dispersal (Campos and Ojeda 1997; Juan et al. 2006), the transmission of rabies disease (Chautan et al. 2000), and its impact on game species (Beja et al. 2009). In Spain, the red fox is a game species with annual hunting quotas. However, the quota implementation follows non-scientific criteria based on abundance estimations. Also, the lack of selective techniques for fox control may harm other endangered carnivores without an effective control of fox abundance (Virgós and Travaini 2005), which highlights the importance of the species for the carnivore guild and associated conservation conflicts.
Using red fox as model species, we tested the following questions: (1) are both methods correlated in detecting the same minimum NI? (2) is the minimum NI index correlated with red fox abundance estimates by both methods? (3) which method is the most economical for estimating red fox abundance in the same spatial and temporal environment? Our applied aim is to provide methodological information to assist decision-making in research requiring estimates of the relative abundance of mesocarnivores.
Materials and methods
Study area
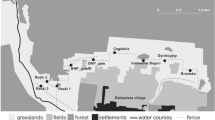
The red fox is continuously distributed in the Autonomous Community of Madrid (Spain). Thus, we selected seven areas in this region for our study: Carabaña (C) and Villarejo de Salvanés (V) in the southeast, La Berzosa (B) and San Mames (SM) in the north and Robledo (R), Quijorna (Q) and Pelayos de la Presa (P) in the southwest (Fig. 1).

Sampling areas, cameras, and trail distribution in the study areas. Carabaña (C), Villarejo de Salvanés (V), La Berzosa (B), San Mames (SM), Robledo (R), Quijorna (Q) and Pelayos de la presa (P). The dots show the camera locations, and the lines show the faecal trails (red is trail 1; yellow is trail 2, and green is trail 3)
The southeast part of the mesomediterranean floor has a temperature range of 5.7–7.5 °C in winter and 20.7–25.2ºC in summer and annual precipitation of around 390 mm per year. Here, rainfed crops, irrigated, and pasture predominate. Oak (Quercus ilex rothundifolia and Quercus coccifera) forests on calcareous soils result in a fragmented landscape characterized by a mixture of open spaces, low vegetation (https://www.comunidad.madrid/servicios/urbanismo-medio-ambiente/parque-regional-sureste), also with a strong human influence (Pascual et al. 2010).
The northern part of our study area lay between the meso- and supramediterranean floors characterized by milder summers and colder winters than the southeastern area; precipitations are also more abundant in this area. The landscape consists of Sclerophyllous vegetation (vegetation with hard leaves, short internodes and leaf orientation parallel or oblique to direct sunlight), oaks with gum rockrose (Cistus spp) and abundant granite boulders. Likewise, scrublands of broom (Cistus spp) and thyme (Thymus vulgaris) with pine woods are present from an altitude of 1400 m a.s.l. The southwest region of Madrid is characterized by a great variety of landscapes, reliefs and plant species, with predominant coniferous forests, especially stone pine (Pinus pinea). On the slopes, there are holm oaks (Quercus ilex), cork oaks (Quercus suber), other oaks (Quercus spp), and junipers (Juniperus communis), which are typical species of Mediterranean forests. Rainfall is abundant throughout the year except in summer, and winters are cold with occasional frosts and snowfalls.
The sampling period took place in 2017 (B, C, SM and R) and 2018 (V, Q and P) to cover different seasons: B, C and P in summer, V and Q in spring, SM in autumn, and R in winter. In this way, differences in detectability and marking that depend on seasonality were avoided (Ralls et al. 2010).
Camera trap** sampling and faeces collection sampling
At each site, we placed eight to ten cameras (Scoutguard SG562C LED White model), spaced approximately 450–600 m apart, covering as large an area as possible to maximize the number of individuals photographed and reduce the likelihood of unsampled foxes (similar to Sarmento et al. 2009). We used ArcGIS 10.2 (ESRI Inc., Redlands, California, USA) to create a minimum convex polygon (MCP) using the camera locations. We placed sardines and a commercial lure (HAGOPUR® Premium Attractant Fox) at the camera-trap sites to increase the probability of detection of foxes (Heinlein et al. 2020; Sebastián-González et al. 2020). The cameras operated for 35 days and were checked every seven days to replenish baits, collect the photographs and check the battery (see Martin-Garcia et al. 2022 for additional information on the photographic sampling design).
We collected faeces in 21 trails of 1 km distributed in seven areas (i.e. three trails per area) (Supplementary Table S1). We cleaned each trail on the first day of camera placement to ensure an exclusive collection of faeces deposited within the sampling period. After that, each trail was sampled every two weeks three times during camera placement to increase the probability of detecting faeces from all individuals in the population. Trails were also inspected the day after the cameras were checked. We chose trails based on proximity to the camera placement areas to increase the probability of detecting the same individuals by both methods (Fig. 1). Fresh faeces that could potentially belong to foxes due to morphology and odour were collected by the same operators. All the operators had broad experience in recognizing carnivore faeces. We placed faeces in 96% alcohol for the first 12 h and then stored them on silica gels at 2–4ºC until they were processed (Nsubuga et al. 2004).
DNA extraction and amplification
We extracted DNA from the faeces using a QIAamp DNA Stool Kit (Qiagen) following the manufacturer´s protocol. DNA purity and quantification were determined with a NanoDrop® 2000 spectrophotometer and Qubit® 3.0 fluorometer Quantitation Kit (Invitrogen™). For species verification, we amplified a short region (120 bp) of the mitochondrial (mt) DNA “ND1” gene. Samples were BLAST using the BLASTx option at the NCBI platform to identify foxes. PCR reactions were performed in a total volume of 20 µL (the protocol for species verification is explained in detail in Supplementary material S1). All amplifications were carried out using filter tips in separate rooms (pre- and post- PCR), and negative controls/blanks were included in all amplifications to avoid contamination. PCR products were run and visualized on a 1.5% agarose gel using gelgreen (BIOTIUM). PCR products were sequenced at Macrogen (Netherlands).
We used 16 autosomal microsatellites loci of different canid species (Canis familiaris, Canis lupus and Vulpes vulpes) to genotype the individuals. The 16 autosomal microsatellites markers were: DB1, DB3 (Holmes et al. 1993); CXX173 (u173), CXX225 (u225), CXX 109 (u109) (Ostrander et al. 1993); c2168. CPH9, CPH1 (Fredholm and Wintero 1995); FH2054 (CXX/c2054), FH2001(CXX/c2001), c2168, c2140, c2004 (Francisco et al. 1996); REN105L 03, INU030 (Sacks et al. 2011) and PEZ03 (Perkin-Elmer, Zoogen). Another set of ten microsatellite loci was unsuccessfully tested (Supplementary Table S2 ). Microsatellites were multiplexed in combinations with similar melting temperatures. We included from two to seven microsatellites in each multiplexed combination (Mplex). For PCR amplification, we followed the two-step multiplexing touch-down approach modified from Arandjelovic et al. 2009 (Supplementary Tables S3 and S4). For the Mplex, microsatellites were amplified by combinations, all in the same PCR reaction (Supplementary Table S3). The amplification was performed in a volume of 20 µL using DreamTaq polymerase (Thermo Scientific). The Mplex was diluted to 1:100 for the second PCR reaction. For the second PCR amplification, the Singleplex (Splex), we used 1 µL of 1:100 diluted multiplex (Mplex) and each microsatellite was amplified independently (see Supplementary Table S4). Splex PCR amplification was performed in a final volume of 10 µL. PCR conditions for the Mplex and the Splex are summarized in Supplementary Tables S3 and S4. The PCR products from the Splex were then diluted in water (1:10), mixed 1 µL with 9.8 µL EDTA (0.1 M) and 0.2 µL of size standard (GeneScanTM, 600 LIZ®, Thermo Scientific) and run on 3730xl DNA Analyzer (Applied Biosystems ™). Samples were genotyped using GeneMapper® Software 5 (Applied Biosystems ™).
Photo-identification and microsatellite genoty**
Individuals were identified based on traits such as body size, age range and, the appearance of the tail, spotting at specific points and other diagnostic features. We followed Sarmento et al. (2009) and Dorning and Harri (2019) to select traits to assist identification (Table 1; see also Fig. 2 for examples of individual identification). We did not account for seasonal changes in coat for each individual. The sampling time was not long enough to appreciate these seasonal differences, and we did not repeat sampling in the same area during different seasons.

Examples of identification of individual foxes by camera trap (a) Individual with two spots at the base of the snout and tail with thick fur. (b) Individual with thin tail and no fur. (c) Individual with a distinct fur pattern on the shoulder, base of the tail, paw and under the neck
Cubs were not individually identifiable due to their juvenile fur lacking sufficient distinctive features. However, juveniles tend to move together, so we decided to consider the maximum number of juveniles appearing together in the same photo as the minimum number of juveniles in the population and include it in the study.
We reviewed photos by a second observer to control for photo-identification bias and reduce overestimation (Foster and Harmsen 2012; Ferreras et al. 2017; Johansson et al. 2020). A third observer re-analyzed fox photographs when the first and second observers disagreed on the number of foxes identified. We considered the minimum number of individuals as the mean of the number of individuals identified by each observer. We also estimated the standard error and confidence intervals of the number of individuals photo-identified.
In microsatellite genoty**, allele sizes were manually scored relative to an internal size standard (GeneScan GS600LIZ) in GeneMapper version 5 (Applied Biosystems ™). Each locus was amplified three times to minimise genoty** errors. Genotypes were accepted as reliable if: (1) a heterozygote was observed at least twice in two independent reactions, and/or (2) a homozygote was observed at least twice independently and failed amplification of the third replicate (Taberlet et al. 1996; Frantz et al. 2003; Flagstad et al. 2004). We defined ambiguous genotypes as (1) genotypes with only one amplified replicate, (2) genotypes that failed all replicates, (3) genotypes that generated different alleles in each replicate, or (4) genotypes with two identical homozygous replicates and a different third. These genotypes were annotated as missing alleles (000000) for the corresponding marker.
We used FreeNA program to test the null allele frequency (Chapuis and Estoup 2009). Gimlet 3.4 (Valière 2003) was used to evaluate heterogeneity observed (Ho) and expected (He); Hardy-Weinberg equilibrium (H-W equilibrium) and probability of identity (PID) to evaluate the reliability of the microsatellites used. The PID among genotypes (Kalinowski et al. 2007) is the most widely used statistical method to quantify the power or ability of molecular markers to distinguish between two individuals. We tested PID, PID (sib), multi-locus PID and Multi-locus PID (sib). The PID is the probability that a single unrelated individual has this genotype (individuals are randomly mated); PID (sib) is the probability that a single full sibling has this genotype (sister-sibling only population), and the multi-locus PID is the PID calculated over several loci by sequentially multiplying the PID value over the loci (considering that the loci are independent) (Scandura et al. 2001). The multi-locus PID (sib) is the PID (sib) calculated over several loci. We considered that a multi-locus PID (sib) less than 0.01 (Mills et al. 2000) or a multi-locus PID between 0.001 and 0.0001 was sufficiently sensitive for identification and avoided underestimation (Waits et al. 2001). The presence of null alleles was checked with the FreeNa program. We also tested the effect of removing markers with a null allele frequency above 0.30 (Dakin and Avise 2004; Huang et al. 2016).
We used Cervus 3.0.7 (Kalinowski et al. 2007) and Gimlet 3.4 to create genotype profiles for all the samples and test the consensus in the number of individuals provided by both programs. Cervus 3.0.7 identifies samples with identical genotypes for the specified number of loci. From our set of 16 microsatellites, we scored individuals as the same if they had identical genotypes for at least eight or more common loci. This grou** method with matching samples ensures a conservative number of identified individuals by minimizing individuals created through erroneous, multi-locus genotypes (Mondol et al. 2009). Also, we allowed a maximum of two mismatching loci to control genoty** errors and increase success in genotype assignment (Kalinowski et al. 2007). We used Gimlet 3.4 to reconstruct the consensus genotypes. Finally, we tested the genotype reassortment function with the assumption that missing alleles are considered distinctive alleles.
Model selection: abundance estimation
We ran N-mixture models to estimate fox abundance from camera trap** without individual identification and accounting for the influence of imperfect detection. We used unmarked package (Fiske and Chandler 2011) in R software (R Core Team 2022) using the pcount function. This function estimates abundance in a hierarchical model. The actual abundance is estimated from the local variation of abundance (λ) at i sites using j temporal counts controlling for detection probability. There are two linked processes for estimating abundance (Kéry and Schaub 2012):
a) Abundance process (λ): Fitted by a Poisson distribution with mean \(\lambda\) and the variation of local abundance at site i
b) Observation process (\(p\)): Fitted by a binomial distribution of the observed counts (\({y}_{i,j}\)) of individuals at each site (\(i\)) in each temporal replicate (\(j\)) with a probability of detection.
We counted each independent fox trap** event in each camera-trap per occasion (24 h). We considered more than one fox capture per occasion when we detected several foxes together in the same independent fox capture event. We first ranked three models to study abundance and detection probability among sampling areas: (1) constant detection probability among areas with a variation of abundance estimates among areas (i.e. \(p\) (.) ~ \(\lambda\) (site)), (2) variation in detection probability and variation of abundance estimates among areas (i.e. \(p\) (site) ~ λ (site)), and (3) variation in detection probability among areas with constant abundance estimates among areas (i.e. \(p\) (site) ~ λ (.)). We compared the performance of the Poisson, zero-inflated Poisson, and negative binomial distributions for each model. For model selection, we used Akaike’s Information Criterion (AIC) (Akaike 1974) corrected for small sample size (AICc) (Burnham and Anderson 2002). We run a chi-square test in Nmix.gof.test function of package AICcmodavg (Mazerolle and Mazerolle 2017) to assess the goodness-of-fit and overdispersion of the selected model. We then estimated the posterior distribution of detection and abundance (\(\lambda\)) using empirical Bayes random effects (ranef) methods from the unmarked package. We used a parametric bootstrap approach with a simulation of 5000 bootstrap samples for each fit assessment. We obtained the mean abundance of each area, the standard error and the confidence interval.
Pearson correlation
We used a Pearson’s correlation test to quantify the proportionality and similarity between (a) minimum photo-identified and genotyped NI, (b) minimum photo-identified NI and abundance estimates, (c) minimum genotyped NI and abundance estimates. We performed a logarithmic transformation when data were not normally distributed. We followed Prion’s classification to establish correlation ranges (Prion and Haerling 2014).
Results
Individual identification: photo-identification and microsatellite genoty**
We obtained 309 photos of foxes. We identified a total of 23.66 individuals with a mean of 3.38 individuals per area (Table 2). Indentified foxes per each observer were described in Supplementary Table S5.
We collected 77 faeces along the trails (Table 2). According to the molecular species identification, mtDNA sequence (“ND1” gene) identified 69 samples of Vulpes vulpes. Based on GIMLET analyses, six samples (three from C and three from Q) could not be assigned to a specific genotype and were eliminated from the analyses, resulting in 63 faecal genotypes.
Consensus genotypes of sample types with 16 microsatellite loci (N = 63) revealed the presence of 19 individuals (Table 2). All loci included in the study were polymorphic. We found 3–12 alleles per locus and an average of 7.06 annotated alleles per locus. The mean allelic dropout rate was 24% (loci = 16; SD = 0.149) among loci and 35% (samples = 69; SD = 0.235) among samples. The single locus probabilities were combined to obtain the total probability over the 16 loci, assuming the independence of different loci. Results of Ho, He and H-W equilibrium of 16 microsatellites are shown in Supplementary Table S6. The different identity probabilities for the 16 microsatellites loci were multi-locus PID = 1.98 × 10–14 and multi-locus PID (sib) = 3.43 × 10− 06. Based on the identified probability, the 16 loci considered were sufficient to distinguish with 99% certainty between sibling red foxes (Supplementary Table S6 ). We obtained null allele values of less than 0.30 in almost all loci, except for INU030, which had a null allele frequency of 0.33 (Supplementary Table S6). We repeated the analyses after removing INU030 and obtained the same results regarding the same number of individuals and sample genotypes. However, the multi-locus PID increased slightly to 1.16 × 10–13. Therefore, we decided to retain this microsatellite in subsequent analyses (Huang et al. 2016). The number of individuals identified was consistent between the two software tools used (Cervus 3.0 and Gimlet 3.4), with a single exception in the P location population, where we detected two individuals by Gimlet 3.4 but could not obtain results with Cervus 3.4. The number of assigned faeces to each individual is described in Supplementary Table S5.
Model selection: abundance estimation
Model selection resulted in two top-ranked models (lowest AICc). The model with lower AICc indicated a constant detection probability and variation in abundance estimation between areas. The second model maintained a constant abundance estimate and a variable detection probability between areas (Table 3).
In both models, the Poisson distribution was more supported than the zero-inflated Poisson and negative binomial. Although the two top-ranked models were close (i.e. ∆AICc < 2), we decided to select the first top-ranked model because it explained our predictions for estimating abundance in each area (Table 3) to study the relationship between abundance estimates and the minimum NI between areas. This model yielded a detection probability of 0.03 (S.E = 0.01; intercept = -3.36; CI [-4.017, -2.694]). The Nmix.gof.test with 100 bootstrapped samples indicated that the selected model fit well (χ2 = 2167.27; p-value = 0.37) with no evidence of overdispersion (c-hat = 1.01) (i.e. observed test statistic divided by the mean of the simulated test statistics).
Pearson correlation
Genotypes for fox NI, photo-identified data of fox NI and abundance data showed a normal distribution (Table 4). We found no significant correlation between the minimum NI derived from photo-identification and faecal genoty** (n = 7; |r| = -0.07; p-value = 0.87), while photo-identified minimum NI showed a strong positive (Prion and Haerling 2014) but non-significant correlation with abundance estimates (n = 7; |r| = 0.68; p-value = 0.08). In contrast, minimum NI from faecal genoty** and abundance estimates were weakly (Prion and Haerling 2014) and non-significantly correlated (n = 7; |r| = − 0.32; p-value = 0.48).
Discussion
Evaluating multiple sampling methods in monitoring carnivore populations is essential for establishing effective conservation strategies (Caughley and Sinclair 1994; Sadlier et al. 2004; Barea-Azcón et al. 2007). We used camera trap** and DNA faecal genoty** to estimate the minimum number of foxes. We estimated fox abundance by implementing N-mixture models to assess and compare the relationship between abundance and minimum NI provided by camera trap** and DNA faecal sampling. First, we found that the estimation of minimum NI provided by camera trap** was slightly higher than that of DNA faecal genoty**. Second, there are indications that areas with more NI identified were those with higher fox abundance, following a positive relationship between abundance and NI detected by camera-trap**. However, we also found a non-significant negative relationship between NI detected by faecal DNA and abundance estimates.
The comparison of camera trap** and DNA faecal sampling methods showed a slight variation in the number of the identified foxes. We identified three more individuals with camera-traps than DNA faecal sampling (minimum NI was identical in Carabaña and Pelayos). However, despite this slight variation, we found no significant correlation between the minimum NI calculated from photo-identified and faecal genotyped. Comparisons between both non-invasive sampling methods to estimate the population abundance of carnivore species are relatively common in the literature (Mondol et al. 2009 (Panthera tigris); Janečka et al. 2011 (Panthera uncia); Galaverni et al. 2012 (Canis lupus); Velli et al. 2015 (Felis silvestris)). Galaverni et al. (2012) found concordance between the two sampling methods in the number of identified wolves, which supported their complementarity. However, the minimum number of wolves identified was slightly higher with the use of DNA than with the cameras. In particular, Mondol et al. (2009) found non-significant differences between sampling methods in tigers, although they detected three more individuals using camera-traps than DNA methods.
We identified fewer individuals using DNA faecal sampling than camera trap** (especially in San Mamés and La Berzosa). This difference could be explained by limitations in the faeces sampling design due to the use of faeces for communication between foxes. Carnivores mark their home ranges and territories with faeces deposited in a non-random distribution (Kruuk 1978; Macdonald 1980; Gorman 1990; Soler et al. 2009). Accounting for this behaviour, we chose random but well-delimited paths (trails and roads) that covered the entire area sampled by cameras. Fox faeces often mark the boundary of territories or sites with critical resources (Macdonald 1985; Barja et al. 2001). Because resource and territory marking occurs unevenly between individuals, marks of specific individuals in a given area may be over-represented (Gorman and Trowbridge 1989). Moreover, defecation rates differ between individuals (Cavallini 1994), particularly between males and females, adults, and juveniles (Goszczyński 1990; Peterson et al. 2002; Ralls et al. 2010; Fawcett et al. 2012), which can influence the number of individuals detected. Defecation rate also vary according to season and diet (Andelt and Andelt 1984; Goszczyński 1990). Other effects, such as changes in marking behaviour caused by the spatial distribution of roads (Vilà et al. 1994; Barja and List 2014; Zaman et al. 2019) may also lead to increased non-uniform distribution of faeces. In our study area of Villarejo, in contrast to other locations, the number of individuals identified by DNA in faeces was higher than using cameras. These differences suggest some limitations of the camera trap** method. Although 32 photos were obtained for identifying individuals, we obtained 15 unidentified photos of either new or previously detected individuals. Camera traps could alter red fox behaviour, specially in females due to the sounds and flashes they make (Meek et al. 2014). Predation risk or anthropogenic disturbance (Lucherini et al. 1995) might also increase trap avoidance, thus influencing individual detection. These potential behavioural changes might impact the more elusive individuals more effusively, with the consequent bias in their detection.
Although we found a non-significant correlation between the genotyped and photo-identified minimum NI, the minimum photo-identified NI and the abundance estimation showed a positive relationship. This result suggests the possibility of using NI as a straightforward index to explain variations in species abundance. We validated individual red fox photo-identification according to previous studies (Sarmento et al. 2009; Dorning and Harris 2019 but see, Güthlin et al. 2014) to obtain a minimum NI. However, we recommend at least three observers to reduce the bias in over- or under-estimating the number of individuals (Foster and Harmsen 2012; Ferreras et al. 2017; Johansson et al. 2020). Photo identification also has limitations for juveniles or cubs. Identifying juveniles becomes difficult when individuals are alone. Therefore, we consider the minimum number of juveniles as the total number of juveniles together in the same photo. However, we acknowledge this approach might be susceptible to underestimating juvenile populations because we considered only the minimum number of juveniles. Nevertheless, we assume that adding juveniles to the study generated less bias when comparing the minimum NI identified and the abundance estimated by both sampling methods. The identification of individuals has become a standard method for the abundance estimation of animal populations, such as using the Spatial Capture-Recapture (SCR) method (Efford 2014; Royle et al. 2014; Rodgers et al. 2014; Wegge et al. 2019). However, CR methods for abundance estimates require a sufficient number of recaptures to obtain accurate estimates (Otis et al. 1978). SCR approaches need at least 20–25 recaptures, including spatial recaptures to correctly describe the movement (Efford et al. 2004), or alternatively using movement data from telemetry tagged individuals (Jimenez et al. 2019) to improve the estimate. Due to the data limitations, we used N-mixture models to estimate fox abundance.
The N-mixture model estimates abundance using count data without needing individual identification or reference to the effective trap** area. Other studies focused on the reliability of the N-mixture models to estimate abundance. Basile et al. (2016) found that N-mixture models and SCR methods yielded a similar estimation of the abundance of the short-toed treecreeper (Certhia brachydactyla). Ficetola et al. (2018) obtained limited differences between N-mixture models and capture-mark-recapture to estimate the abundance of small vertebrates. Also, to avoid violating assumptions of N-mixture caused by double counting of a single sampling occasion (Link et al. 2018), we only considered more than one fox capture per occasion (24 h) when we detected several foxes together in the same capture event. Our results between photo-identified NI and abundance showed that NI results are helpful when the minimum number of identified individuals in the population is not sufficient, or the number of captures of identified individuals is not enough to produce reliable abundance estimates (Otis et al. 1978). NI may also be recommended when sufficient temporal and spatial replicates are unavailable or the assumptions of N-mixture models are not met (e.g. independence in the case of gregarious animals). Thus, Martin-Garcia et al. (2022) found that the minimum photo-identified NI might not be biased by detection probability, thus obtaining the same predictors as the N-Mixture models to explain abundance patterns.
We found no significant correlation between minimum NI genotyped and abundance. Including more faeces could help identify a potential relationship between the minimum number of individuals identified by DNA faeces sampling, NI photo-identified, and abundance (Wegge et al. 2019; Lindsø et al. 2022). Future research should include random transects out of existing trails to increase the number of faeces and detections of individuals. Implementing a random transect design that covers many landscapes with different compositions and configurations (Güthlin et al. 2012) could reduce the bias caused by some individuals marking more intensively along the trails. In this vein, we could better refine and compare the relationship between estimated abundances of faecal DNA and camera trap** sampling methods (Rodgers et al. 2014) and between NI genotypes and abundance. In addition, new state-of-the-art genomic approaches based on SNPs approach (e.g. RAD-seq) may increase the accuracy of DNA amplification decreasing the loss of samples and consequently improving abundance estimates (Andrews et al. 2016; De Barba et al. 2017; Erwin et al. 2021). Another timely method is the SNP genoty** method based on high-throughput real-time PCR technique known as Dynamic Array™ by Fluidigm® that is mainly used for degraded samples such as faeces and ancient DNA studies samples (Kraus et al. 2015).
In our experience, using camera trap** was cheaper than DNA sampling methods to study red foxes. Our budget for the camera trap** method was 2756 euros (including cameras, baits and placement in the study areas) compared to 7500 euros for the faecal DNA method (including fieldwork, sample shipment and protocol optimization). Several aspects should be considered in terms of budget. Firstly, a small pilot optimization study beforehand could help to reduce costs in future analyses. The optimization process with fewer samples is helpful to check the protocol used and obtain preliminary results avoiding using all the valuable samples. Secondly, using other less costly genetic techniques. Single nucleotide polymorphism (SNPs) is frequently used as a new genoty** method for individual and sex identification (Parker et al. 2021; Buchalski et al. 2022; Lopez-Bao et al. 2020). To date, develo** a SNP panel for individual identification is considered an efficient and cost-effective method to simultaneously genotype hundreds of individuals (Carroll et al. 2018). Lastly, researchers should consider an extra budget for the replacement of the cameras in case of loss or failure during the study. Consequently, the budget for camera trap** may increase depending on the number of cameras needed for our research.
Our research highlights the importance of correctly selecting sampling methods for abundance studies. Researchers can adjust the broad choices in sampling methods to their available funds and logistics. Different methods can perform differently and provide different results; thus, it is required to identify first the costs and limitations of the potential techniques for our specific research objectives and the species to study. Our results suggest the minimum photo-identified NI is a reliable index for studying abundance variation when other methods are unavailable. In contrast, it is necessary to improve the methods of faeces sampling to estimate population size and to explore its relationship with camera trap data. Sampling designs with transects away from existing trails will increase the probability of finding more faeces. We should mention that both methods were compared to study abundance over a short period. On the other hand, DNA sampling in faeces could be useful to identify individuals over a longer period, over years and seasons when this would be very difficult with photos (Bellemain et al. 2005). In addition, assessments of genetic diversity, population substructure, gene flow, paternity, and heritability are easy to evaluate with DNA stool genoty** (Pilot et al. 2014; Zanin et al. 2016). However, this was not the main objective of this study. Regarding our model species, the red fox, camera trap** methods can be a reference for future red fox management actions. In our study, the validity of using the camera trap** method to estimate fox population abundance is also motivated by its lower cost when compared to the faecal DNA genotyped method. However, further research on the cost-effectiveness of new genetic methods is encouraged.
Data Availability
All data generated or analyzed during this study are included in this published article and supplementary information files.
References
Andelt WF, Andelt SH (1984) Diet bias in scat deposition-rate surveys of coyote density. Wildl Soc Bull (1973–2006) 12(1):74–77
Anderson DR (2003) Response to Engeman: index values rarely constitute reliable information. Wildl Soc Bull 31(1):288–291
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81–92. https://doi.org/10.1038/nrg.2015.28
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Autom Control 19(6):716–723. https://doi.org/10.1109/TAC.1974.1100705
Arandjelovic M, Guschanski K, Schubert G, Harris TR, Thalmann O, Siedel H, Vigilant L (2009) Two-step multiplex polymerase chain reaction improves the speed and accuracy of genoty** using DNA from noninvasive and museum samples. Mol Ecol Resour 9(1):28–36. https://doi.org/10.1111/j.1755-0998.2008.02387.x
Balme GA, Hunter LT, Slotow R (2009) Evaluating methods for counting cryptic carnivores. J Wildl Manag 73(3):433–441. https://doi.org/10.2193/2007-368
Barea-Azcón JM, Virgós E, Ballesteros-Duperon E, Moleón M, Chirosa M (2007) Surveying carnivores at large spatial scales: a comparison of four broad-applied methods. Biodivers Conserv 16(4):1213–1230. https://doi.org/10.1007/s10531-006-9114-x
Barja I, De Miguel FJ, Bárcena F (2001) Distribución espacial de los excrementos de zorro rojo (Vulpes vulpes, Linnaeus 1758) en los Montes do Invernadeiro (Ourense). Galemys 13:171–178
Barja I, List R (2014) The role of spatial distribution of faeces in coyote scent marking behaviour. Pol J Ecol 62(2):373–384. https://doi.org/10.3161/104.062.0215
Basile M, Balestrieri R, Posillico M, Mancinelli A, Altea T, Matteucci G (2016) Measuring bird abundance—a comparison of methodologies between capture/recapture and audio-visual surveys. Avocetta 40:55–61
Beja P, Gordinho L, Reino L, Loureiro F, Santos-Reis M, Borralho R (2009) Predator abundance in relation to small game management in southern Portugal: conservation implications. Eur J Wildl Res 55(3):227–238. https://doi.org/10.1007/s10344-008-0236-1
Bellemain EVA, Swenson JE, Tallmon D, Brunberg S, Taberlet P (2005) Estimating population size of elusive animals with DNA from hunter-collected feces: four methods for brown bears. Conserv Biol 19(1):150–161. https://doi.org/10.1111/j.1523-1739.2005.00549.x
Buchalski MR, Benjamin N, Sacks KD, Ahrens, Kyle D, Gustafson JL, Rudd HB, Ernest, Justin A (2022) Dellinger. “Development of a 95 SNP panel to individually genotype mountain lions (Puma concolor) for microfluidic and other genoty** platforms. Conserv Genet Resour ( 1–4. https://doi.org/10.1007/s12686-022-01255-6
Burnham KP, Anderson DR (2002) A Practical Information-Theoretic Approach. Model Selection and Multimodel Inference, 2nd ed. Springer, New York, p. 2
Campos CM, Ojeda RA (1997) Dispersal and germination ofProsopis flexuosa (Fabaceae) seeds by desert mammals in Argentina. J Arid Environ 35(4):707–714. https://doi.org/10.1006/jare.1996.0196
Carroll EL, Bruford MW, DeWoody JA, Leroy G, Strand A, Waits L, Wang J (2018) Genetic and genomic monitoring with minimally invasive sampling methods.698. Evol Appl 11(7):1094–1119. https://doi.org/10.1111/eva.12600
Caughley G, Sinclair ARE (1994) Wildlife Ecology and Management. Blackwell Science: Melbourne)
Chapuis MP, Estoup A (2009) FreeNA.
Chautan M, Pontier D, Artois M (2000) Role of rabies in recent demographic changes in red fox (Vulpes vulpes) populations in Europe. Mammalia 64(4):391–410
Cavallini P (1994) Faeces count as an index of fox abundance. Acta Theriol 39:417–417
Costa A, Romano A, Salvidio S (2020) Reliability of multinomial N-mixture models for estimating abundance of small terrestrial vertebrates. Biodivers Conserv 29(9):2951–2965. https://doi.org/10.1007/s10531-020-02006-5
Dakin EE, Avise JC (2004) Microsatellite null alleles in parentage analysis. Heredity 93(5):504–509. https://doi.org/10.1038/sj.hdy.6800545
De Barba M, Miquel C, Lobréaux S, Quenette PY, Swenson JE, Taberlet P (2017) High-throughput microsatellite genoty** in ecology: improved accuracy, efficiency, standardization and success with low-quantity and degraded DNA. Mol Ecol Resour 17:492–507. https://doi.org/10.1111/1755-0998.12594
Dorning J, Harris S (2019) The challenges of recognising individuals with few distinguishing features: identifying red foxes Vulpes vulpes from camera-trap photos.PloS one, 14(5), e0216531. https://doi.org/10.1371/journal.pone.0216531
Efford MG (2004) Density estimation in live-trap** studies. Oikos 106:598–610. https://doi.org/10.1111/j.0030-1299.2004.13043.x
Efford MG, Dawson DK, Robbins CS (2004) DENSITY: Software for analysing capture-recapture data from passive detector arrays. Anim Biodivers Conserv 27:217–228
Erwin JA, Robert R, Fitak, Culver M (2021) PumaPlex100: an expanded tool for puma SNP genoty** with low-yield DNA. Conserv Genet Resour 13(3):341–343. https://doi.org/10.1007/s12686-021-01206-7
Ferreras P, Jiménez J, Tobajas J, Ramos S, Descalzo E, Mateo R (2017) Estimating red fox (Vulpes vulpes) abundance with spatial mark-resight models and camera traps: the effects of tagging, observer and individual recognition
Ficetola GF, Barzaghi B, Melotto A, Muraro M, Lunghi E, Canedoli C, Lo Parrino E, Nanni V, Silva-Rocha I, Urso A, Carretero MA, Salvi D, Scali S, Scarì G, Pennati R, Andreone F, Manenti R (2018) N-mixture models reliably estimate the abundance of small vertebrates. Sci Rep 8(1). https://doi.org/10.1038/s41598-018-28432-8
Fiske I, Chandler R (2011) Unmarked: an R package for fitting hierarchical models of wildlife occurrence and abundance. J Stat Softw 43(10):1–23. https://doi.org/10.18637/jss.v043.i10
Flagstad Ø, Hedmark EVA, Landa A, Brøseth H, Persson J, Andersen R, …, Ellegren H (2004) Colonization history and noninvasive monitoring of a reestablished wolverine population. Conserv Biol 18(3):676–688. https://doi.org/10.1111/j.1523-1739.2004.00328.x-i1
Foster RJ, Harmsen BJ (2012) A critique of density estimation from camera-trap data. J Wildl Manag 76(2):224–236. https://doi.org/10.1002/jwmg.275
Francisco LV, Langsten AA, Mellersh CS, Neal CL, Ostrander EA (1996) A class of highly polymorphic tetranucleotide repeats for canine genetic map**. Mamm Genome 7(5):359–362. https://doi.org/10.1007/s003359900104
Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, Delahay RJ, Burke T (2003) Reliable microsatellite genoty** of the eurasian badger (Meles meles) using faecal DNA. Mol Ecol 12(6):1649–1661. https://doi.org/10.1046/j.1365-294X.2003.01848.x
Fredholm M, Winterø AK (1995) Variation of short tandem repeats within and between species belonging to the Canidae family. Mamm Genome 6(1):11–18. https://doi.org/10.1007/BF00350887
Gaidet-Drapier N, Fritz H, Bourgarel M, Renaud PC, Poilecot P, Chardonnet P, …, Le Bel S (2006) Cost and efficiency of large mammal census techniques: comparison of methods for a participatory approach in a communal area, Zimbabwe. Biodivers Conserv 15(2):735–754. https://doi.org/10.1007/s10531-004-1063-7
Galaverni M, Palumbo D, Fabbri E, Caniglia R, Greco C, Randi E (2012) Monitoring wolves (Canis lupus) by non-invasive genetics and camera trap**: a small-scale pilot study. Eur J Wildl Res 58(1):47–58. https://doi.org/10.1007/s10344-011-0539-5
Gittleman JL, Funk SM, MacDonald DW, Wayne RK (eds) (2001) Carnivore conservation, vol 5. Cambridge University Press, Cambridge
Gompper ME, Kays RW, Ray JC, Lapoint SD, Bogan DA, Cryan JR (2006) A comparison of noninvasive techniques to survey carnivore communities in northeastern North America. Wildl Soc Bull 34(4):1142–1151. https://doi.org/10.2193/0091-7648(2006)34[1142:ACONTT]2.0.CO;2
Gorman ML (1990) Scent marking strategies in mammals. Rev Suisse Zool 97:3–30
Gorman ML, Trowbridge BJ (1989) The role of odor in the social lives of carnivores. Carnivore behaviour, ecology, and evolution. Springer, Boston, MA, pp 57–88. https://doi.org/10.1007/978-1-4757-4716-4_3
Goszczyński J (1990) Scent marking by red foxes in Central Poland during the winter season. ActaTheriologica 35(1–2):7–16
Güthlin D, Kröschel M, Küchenhoff H, Storch I (2012) Faecal sampling along trails: a questionable standard for estimating red fox Vulpes vulpes abundance. Wildl Biology 18(4):374–382. https://doi.org/10.2981/11-065
Güthlin D, Storch I, Küchenhoff H (2014) Is it possible to individually identify red foxes from photographs? Wildl Soc Bull 38(1):205–210. https://doi.org/10.1002/wsb.377
Heinlein BW, Urbanek RE, Olfenbuttel C, Dukes CG (2020) Effects of different attractants and human scent on mesocarnivore detection at camera traps. Wildl Res 47(4):338–348. https://doi.org/10.1071/WR19117
Holmes NG, Humphreys SJ, Binns MM, Holliman A, Curtis R, Mellersh CS, Sampson I (1993) Isolation and characterization of microsatellites from the canine genome. Anim Genet 24(4):289–292. https://doi.org/10.1111/j.1365-2052.1993.tb00313.x
Huang K, Ritland K, Dunn DW, Qi X, Guo S, Li B (2016) Estimating relatedness in the presence of null alleles. Genetics 202(1):247–260. https://doi.org/10.2193/0091-7648(2006)34[69:EBAUAT]2.0.CO;2
Janečka JE, Munkhtsog B, Jackson RM, Naranbaatar G, Mallon DP, Murphy WJ (2011) Comparison of noninvasive genetic and camera-trap** techniques for surveying snow leopards. J Mammal 92(4):771–783. https://doi.org/10.1644/10-MAMM-A-036.1
Jackson RM, Roe JD, Wangchuk R, Hunter DO (2006) Estimating snow leopard population abundance using photography and capture-recapture techniques. Wildl Soc Bull 34(3):772–781. https://doi.org/10.2193/0091-7648(2006)34[772:ESLPAU]2.0.CO;2
Jiménez J, Nuñez-Arjona JC, Rueda C, González LM, García-Domínguez F, Muñoz-Igualada J, López-Bao JV (2017) Estimating carnivore community structures. Sci Rep 7:1–10. https://doi.org/10.1038/srep41036
Jimenez J, Chandler R, Tobajas J, Descalzo E, Mateo R, Ferreras P (2019a) Generalized spatial mark–resight models with incomplete identification: an application to red fox density estimates. Ecol Evol 9(8):4739–4748. https://doi.org/10.1002/ece3.5077
Jiménez J, Nuñez-Arjona JC, Mougeot F, Ferreras P, González LM, García-Domínguez F, Muñoz-Igualada J, Palacios MJ, Pla S, Rueda C, Villaespesa F, Nájera F, Palomares F, López-Bao JV (2019b) Restoring apex predators can reduce mesopredator abundances. Biol Conserv 238:108234. https://doi.org/10.1016/j.biocon.2019.108234
Johansson Ö, Samelius G, Wikberg E, Chapron G, Mishra C, Low M (2020) Identification errors in camera-trap studies result in systematic population overestimation. Sci Rep 10(1):1–10. https://doi.org/10.1038/s41598-020-63367-z
Juan T, Sagrario A, Jesús H, Cristina CM (2006) Red fox (Vulpes vulpes L.) favour seed dispersal, germination and seedling survival of Mediterranean Hackberry (Celtis australis L). Acta Oecol 30(1):39–45. https://doi.org/10.1016/j.actao.2006.01.004
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genoty** error increases success in paternity assignment. Mol Ecol 16(5):1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Karanth KU, Nichols JD (1998) Estimation of tiger densities in India using photographic captures and recaptures. Ecology 79(8):2852–2862
Kéry M, Schaub M (2012) Bayesian population analysis using WinBUGS. A hierarchical perspective. Academic Press / Elsevier
Kidwai Z, Jimenez J, Louw CJ, Nel HP, Marshal JP (2019) Using N-mixture models to estimate abundance and temporal trends of black rhinoceros (Diceros bicornis L.) populations from aerial counts. Global Ecol Conserv 19:e00687. https://doi.org/10.1016/j.gecco.2019.e00687
Kraus RH, Vonholdt B, Cocchiararo B, Harms V, Bayerl H, Kühn R, Nowak C (2015) A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Resour 15(2):295–305. https://doi.org/10.1111/1755-0998.12307
Kruuk H (1978) Spatial organization and territorial behaviour of the european badger Meles meles. J Zool 184(1):1–19. https://doi.org/10.1111/j.1469-7998.1978.tb03262.x
Lindsø LK, Dupont P, Rød-Eriksen L, Andersskog IPØ, Ulvund KR, Flagstad Ø, …, Eide NE (2022) Estimating red fox density using non-invasive genetic sampling and spatial capture–recapture modelling. Oecologia 198(1):139–151. https://doi.org/10.1007/s00442-021-05087-3
Link WA, Schofield MR, Barker RJ, Sauer JR (2018) On the robustness of N-mixture models. Ecology 99(7):1547–1551. https://doi.org/10.1002/ecy.2362
Linnell JD, Strand O (2000) Interference interactions, co-existence and conservation of mammalian carnivores. Divers Distrib 6(4):169–176. https://doi.org/10.1046/j.1472-4642.2000.00069.x
Lloyd HG (1980) Habitat requirements of the red fox. InThe red fox. Springer, Dordrecht, pp 7–25
López-Bao J, Vicente R, Godinho R, Gomes Rocha G, Palomero JC, Blanco (2020) Fernando Ballesteros, and José Jiménez. “Consistent bear population DNA-based estimates regardless molecular markers type. " Biol Conserv 248:108651. https://doi.org/10.1016/j.biocon.2020.108651
Lucherini M, Lovari S, Crema G (1995) Habitat use and ranging behaviour of the red fox (Vulpes vulpes) in a Mediterranean rural area: is shelter availability a key factor? J Zool 237(4):577–591. https://doi.org/10.1111/j.1469-7998.1995.tb05016.x
Macdonald DW (1985) The carnivores: order Carnivora. In: Brown RE (ed) Social Odours in Mammals/Eds. Macdonald DW
MacDonald DW (1980) Patterns of scent marking with urine and faeces amongst carnivore communities. In Symposia of the Zoological Society of London (Vol 45(107):e139
Martin-Garcia S, Rodríguez-Recio M, Peragón I, Bueno I, Virgós E (2022) Comparing relative abundance models from different indices, a study case on the red fox. Ecol Ind 137:108778. https://doi.org/10.1016/j.ecolind.2022.108778
Mazerolle MJ, Mazerolle MMJ (2017) Package ‘AICcmodavg’.R package,281
Meek PD, Ballard GA, Fleming PJ, Schaefer M, Williams W, Falzon G (2014) Camera traps can be heard and seen by animals. PLoS ONE 9(10). https://doi.org/10.1371/journal.pone.0110832
Meek PD, Ballard GA, Fleming PJ (2015) The pitfalls of wildlife camera trap** as a survey tool in Australia. Australian Mammalogy 37(1):13–22. https://doi.org/10.1071/AM14023
Mills LS, Citta JJ, Lair KP, Schwartz MK, Tallmon DA (2000) Estimating animal abundance using noninvasive DNA sampling: promise and pitfalls. Ecol Appl 10(1):283–294. https://doi.org/10.1890/1051-0761010[0283:EAAUND]2.0.CO;2
Mondol S, Karanth KU, Kumar NS, Gopalaswamy AM, Andheria A, Ramakrishnan U (2009) Evaluation of non-invasive genetic sampling methods for estimating tiger population size. Biol Conserv 142(10):2350–2360. https://doi.org/10.1016/j.biocon.2009.05.014
Mosquera D, Blake JG, Swing K, Romo D (2016) Ocelot (Leopardus pardalis) density in Eastern Ecuador based on capture–recapture analyses of camera trap data. Neotropical Biodivers 2(1):51–58. https://doi.org/10.1080/23766808.2016.1168593
Murphy MA, Kendall KC, Robinson A, Waits LP (2007) The impact of time and field conditions on brown bear (Ursus arctos) faecal DNA amplification. Conserv Genet 8(5):1219–1224
Nsubuga AM, Robbins MM, Roeder AD, Morin PA, Boesch C, Vigilant L (2004) Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol Ecol 13(7):2089–2094. https://doi.org/10.1111/j.1365-294X.2004.02207.x
O’Connell AF, Nichols JD, Karanth KU (2011) Camera traps in animal ecology: methods and analyses (p. 271). Tokyo: Springer. odours in Mammals. Clarendon Press, Oxford: 619–722
Ostrander EA, Sprague Jr GF, Rine J (1993) Identification and characterization of dinucleotide repeat (CA) n markers for genetic map** in dog. Genomics 16(1):207–213. https://doi.org/10.1006/geno.1993.1160
Otis DL, Burnham KP, White GC, Anderson DR (1978) Statistical inference from capture data on closed animal populations.Wildlife monographs, (62),3–135
Parker LD, Campana MG, Quinta JD, Cypher B, Rivera I, Fleischer RC, Maldonado JE (2021) An efficient noninvasive method for simultaneous species, individual, and sex identification of sympatric Mojave Desert canids via in-solution SNP capture. Authorea Preprints
Pascual Rosa V, Aguilera Benavente F, Rocha P, Gómez Delgado W, M., Bosque Sendra J (2010) Simulación de modelos de crecimiento urbano: métodos de comparación con los mapas reales. In Congreso Nacional de Tecnologías de la Información Geográfica (14. 2010. Sevilla) (2010), p 1.000-1.013. Universidad de Sevilla
Peterson RO, Jacobs AK, Drummer TD, Mech LD, Smith DW (2002) Leadership behaviour in relation to dominance and reproductive status in gray wolves, Canis lupus. Can J Zool 80(8):1405–1412. https://doi.org/10.1139/z02-124
Pilot M, Dąbrowski MJ, Hayrapetyan V, Yavruyan EG, Kopaliani N, Tsingarska E, Bogdanowicz W (2014) Genetic variability of the grey wolf Canis lupus in the Caucasus in comparison with Europe and the Middle East: distinct or intermediary population? PLoS ONE 9(4):e93828. https://doi.org/10.1371/journal.pone.0093828
Pollock KH (1976) Building models of capture-recapture experiments. J Royal Stat Soc Ser D (The Statistician) 25(4):253–259. https://doi.org/10.2307/2988083
Prion S, Haerling KA (2014) Making sense of methods and measurement: Pearson product-moment correlation coefficient. Clin Simul Nurs 11(10):587–588. https://doi.org/10.1016/j.ecns.2014.07.010
Prugh LR, Stoner CJ, Epps CW, Bean WT, Ripple WJ, Laliberte AS, Brashares JS (2009) The rise of the mesopredator. Bioscience 59(9):779–791. https://doi.org/10.1525/bio.2009.59.9.9
Ralls K, Sharma S, Smith DA, BREMNER-HARRISON S, Cypher BL, Maldonado JE (2010) Changes in kit fox defecation patterns during the reproductive season: implications for noninvasive surveys. J Wildl Manag 74(7):1457–1462. https://doi.org/10.1111/j.1937-2817.2010.tb01272.x
R Core Team (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Ritchie EG, Elmhagen B, Glen AS, Letnic M, Ludwig G, McDonald RA (2012) Ecosystem restoration with teeth: what role for predators? Trends Ecol Evol 27(5):265–271. https://doi.org/10.1016/j.tree.2012.01.001
Rodgers TW, Giacalone J, Heske EJ, Janečka JE, Phillips CA, Schooley RL (2014) Comparison of noninvasive genetics and camera trap** for estimating population density of ocelots (Leopardus pardalis) on Barro Colorado Island, Panama. Trop Conserv Sci 7(4):690–705. https://doi.org/10.1177/194008291400700408
Romesburg HC (1981) Wildlife science: gaining reliable knowledge.The Journal of Wildlife Management,293–313
Royle JA (2004) N-mixture models for estimating population size from spatially replicated counts. Biometrics 60(1):108–115. https://doi.org/10.1111/j.0006-.2004.00142.x
Royle JA, Chandler RB, Sollmann R, Gardner B (2014) Spatial capture-recapture. Elsevier, Academic Press, Waltham, Massachusetts
Sacks BN, Moore M, Statham MJ, Wittmer HU (2011) A restricted hybrid zone between native and introduced red fox (Vulpes vulpes) populations suggests reproductive barriers and competitive exclusion. Mol Ecol 20(2):326–341. https://doi.org/10.1111/j.1365-294X.2010.04943.x
Sadlier LM, Webbon CC, Baker PJ, Harris S (2004) Methods of monitoring red foxes Vulpes vulpes and badgers Meles meles: are field signs the answer? Mammal Rev 34(1–2):75–98. https://doi.org/10.1046/j.0305-1838.2003.00029.x
Sarmento P, Cruz J, Eira C, Fonseca C (2009) Evaluation of camera trap** for estimating red fox abundance. J Wildl Manag 73(7):1207–1212. https://doi.org/10.2193/2008-288
Scandura M, Apollonio M, Mattioli L (2001) Recent recovery of the italian wolf population: a genetic investigation using microsatellites. Mammalian Biology 66:321–331
Sebastián-González E, Morales-Reyes Z, Naves-Alegre L, Alemañ CJD, Lima LG, Lima LM, Sánchez- Zapata JA (2020) Which bait should I use? Insights from a camera trap study in a highly diverse cerrado forest. Eur J Wildl Res 66(6):1–8. https://doi.org/10.1007/s10344-020-01439-1
Silveira L, Jacomo AT, Diniz-Filho JAF (2003) Camera trap, line transect census and track surveys: a comparative evaluation. Biol Conserv 114(3):351–355. https://doi.org/10.1016/S0006-3207(03)00063-6
Soisalo MK, Cavalcanti SM (2006) Estimating the density of a jaguar population in the brazilian pantanal using camera-traps and capture–recapture sampling in combination with GPS radio-telemetry. Biol Conserv 129(4):487–496. https://doi.org/10.1016/j.biocon.2005.11.023
Soler L, Lucherini M, Manfredi C, Ciuccio M, Casanave EB (2009) Characteristics of defecation sites of the Geoffroy’s cat Leopardus geoffroyi. Mastozoología Neotropical 16(2):485–489
Sollmann R, Tôrres NM, Furtado MM, de Almeida Jácomo AT, Palomares F, Roques S, Silveira L (2013) Combining camera-trap** and noninvasive genetic data in a spatial capture–recapture framework improves density estimates for the jaguar. Biol Conserv 167:242–247. https://doi.org/10.1016/j.biocon.2013.08.003
Srbek-Araujo AC, Chiarello AG (2005) Is camera-trap** an efficient method for surveying mammals in neotropical forests? A case study in south-eastern Brazil. J Trop Ecol 21(1):121–125
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Bouvet J (1996) Reliable genoty** of samples with very low DNA quantities using PCR. Nucleic Acids Res 24(16):3189–3194
Trolle M, Noss AJ, Lima EDS, Dalponte JC (2007) Camera-trap studies of maned wolf density in the Cerrado and the Pantanal of Brazil. Biodivers Conserv 16(4):1197–1204. https://doi.org/10.1007/978-1-4020-6320-6_24
Valière N (2003) GIMLET v. 1.3. 2 guide. Laboratoire de Biométrie et biologie Evolutive-UMR5558-43, boulevard du 11 novembre 1918-F69622, Villeurbanne, France
Veech JA, Ott JR, Troy JR (2016) Intrinsic heterogeneity in detection probability and its effect on N-mixture models. Methods Ecol Evol 7(9):1019–1028. https://doi.org/10.1111/2041-210X.12566
Velli E, Bologna MA, Silvia C, Ragni B, Randi E (2015) Non-invasive monitoring of the european wildcat (Felis silvestris silvestris Schreber, 1777): comparative analysis of three different monitoring techniques and evaluation of their integration. Eur J Wildl Res 61(5):657–668
Vilà C, Urios V, Castroviejo J (1994) Use of faeces for scent marking in Iberian wolves (Canis lupus). Can J Zool 72(2):374–377. https://doi.org/10.1139/z94-053
Virgós E, Travaini A (2005) Relationship between small-game hunting and carnivore diversity in central Spain. Biodivers Conserv 14(14):3475–3486. https://doi.org/10.1007/s10531-004-0823-8
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10(1):249–256. https://doi.org/10.1046/j.1365-294X.2001.01185.x
Wegge P, Bakke BB, Odden M, Rolstad J (2019) DNA from scats combined with capture recapture modeling: a promising tool for estimating the density of red foxes a pilot study in a boreal forest in southeast Norway. Mammal Res 64(1):147–154. https://doi.org/10.1007/s13364-018-0408-7
Zaman M, Tolhurst B, Zhu M, Heng B, Jiang G (2019) Do Red Foxes (Vulpes vulpes) increase the detectability of Scent Marks by selecting highly conspicuous substrates? J Ethol Anim Sci 2(2):000113
Zanin M, Adrados B, González N, Roques S, Brito D, Chávez C, Palomares F (2016) Gene flow and genetic structure of the puma and jaguar in Mexico. Eur J Wildl Res 62(4):461–469. https://doi.org/10.1007/s10344-016-1019-8
Acknowledgements
We thank all the volunteers who participated in the data collection, especially Javier Hernandez, Ivan Peragón and Tamara Burgos. Special mention to Marina Sanz, Emma Valero and Gunilla Engström for their assistance in the lab. We also thank Katerina Guschanski for her helpful advice, Charles Cong Xu and Frida Lona Durazo for designing the mitochondrial primers, and Jaelle Brealey for hel** with English.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Contributions
SMG: Data collection, laboratory work, and Data Analyses. Conceptualization of the project and the first draft of the manuscript. MCC: designing and supervising molecular lab work, writing and revising the manuscript. MRR: review, edit, and comment on the manuscript. JJ: Data analysis supervisor. JH: Molecular host lab PI and revision of the manuscript. EV: Conceptualization and supervision of the project. Writing and revision of the manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest/Competing interests
We declare no conflicts of interest/competing interests.
Additional information
Communicated By Alison Nazareno.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martin-Garcia, S., Cortazar-Chinarro, M., Rodríguez-Recio, M. et al. Comparing minimum number of individuals and abundance from non-invasive DNA sampling and camera trap** in the red fox (Vulpes vulpes). Biodivers Conserv 32, 1977–1998 (2023). https://doi.org/10.1007/s10531-023-02586-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-023-02586-y