
Abstract
The genetic population structure relationships of Hyalomma (Euhyalomma) lusitanicum in Andalusia (the south of the Iberian Peninsula) were examined using mtDNA sequence data from 887 bp of cytochrome oxidase subunit I (COI) gene. The sequence for the COI region was determined for 84 individuals collected in several localities of Andalusia, and 10 for other localities (i.e., five from Toledo, central Iberian Peninsula, four from Sicily (Italy) and one from Canary Island). Seventeen haplotypes were detected, including 27 polymorphic sites. The number of amino acid substitutions per site from mean diversity calculations for the entire population was 0.017. AMOVA analysis revealed a low gene flow that characterises the genetic population structure of this species in South Iberian Peninsula, with a haplotype diversity (h) value of 0.815. No geographically induced differentiation was observed, and separate evolutionary units were not detected. Our results indicate low genetic diversity across the geographical range of H. lusitanicum tick in Andalusia. Our data do not show any genetic discontinuity between the tick populations studied, including specimens from Canary Island and Sicily (Italy).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyalomma lusitanicum Koch has been included in the ‘anatolicum’ group, together with several species and subspecies (H. anatolicum, H. excavatum and H. franchinii) (Guglielmone et al. 2014). The mature phases of this species, like all other types of subgenus Euhyalomma, parasitize large ungulates, mainly cattle (Apanaskevich 2004). On the Iberian Peninsula, its distribution range is restricted to the southern half (Estrada-Peña et al. 2004; Estrada-Peña and Santos-Silva 2005; Ruiz-Fons et al. 2006). It is also known to occur on the Balearic Island of Menorca (Castellà et al. 2001). Furthermore, the species has been reported from Morocco, Algeria, Italy (including Sicily) and France (Torina et al. 2006, 2010; Santos-Silva 2017). Due to the strategic geographical position, Andalusian H. lusitanicum populations represent a melting pot that connect other Iberian Peninsula populations with North Africa populations as well as with Mediterranean and Atlantic colonized islands with dispersion driven by bird migration and human action (Palomar et al. 2013).
The three-host life cycle of H. lusitanicum has been studied under laboratory conditions (Ouhelli and Pandey 1984; Ouhelli 1994), with larva and nymph stages able to feed on rabbits and cattle, whereas adults engorge on cattle but not on rabbits. Such studies have not been conducted under natural conditions, but the Mediterranean rabbit and various ungulates may be the main hosts for immature and adult H. lusitanicum ticks, respectively (González et al. 2016; Valcárcel et al. 2016).
Several Hyalomma species are susceptible to infection with the highly pathogenic Theileria annulata, the causative agent of tropical theilerosis (Sayin et al. 2003). In the Iberian Peninsula and the Balearic Islands, T. annulata coexists frequently with T. buffeli which is considered as a non-malignan species (Georges et al. 2001; Brígido et al. 2004), likely transmitted by Haemaphysalis spp. ticks (García-Sanmartín et al. 2006). In this region, both H. lusitanicum and H. marginatum has been incriminated in the transmission of T. annulata and Theileria spp. (Habela et al. 1999; Viseras et al. 1999; Almería et al. 2001; Santos-Silva et al. 2011; Gomes et al. 2013; Pereira et al. 2016). In recent times, H. lusitanicum has been implicated as reservoir of Crimean-Congo haemorrhagic fever virus (CCHFV) in central Iberian Peninsula (Estrada-Peña et al. 2012a, b; Negredo et al. 2019).
The wide distribution of CCHFV (genotype Africa IV and Europe V) in the centre and southwest of the Iberian Peninsula in various species of ungulates (red deer, fallow deer and Eurasian wild boar) and associated tick species H. lusitanicum and D. marginatus (Moraga-Fernández et al. 2020) introduces the need to supplement epidemiological studies with studies that highlight the phylogeography of potential vector ticks of this arbovirus in order to establish their population structure and possible dispersal routes in this area. The phylogeny of ticks has been extensively studied to determine the relative positions of genera (Murrell et al. 2001). Mitochondrial genes are among the most widely used markers for phylogenetic studies of animals (Avise 2004). In the case of ticks, mitochondrial DNA (mtDNA) sequences have been extensively used to investigate between populations within species (Mtambo et al. 2007) and the relationships of closely related species (Caporale et al. 1995; Zahler et al. 1995) for their rapid rate of evolution.
Molecular studies focussing on Hyalomma genus are very scarce and partial in the taxonomic and geographical scopes. More recently, the use of mtDNA sequences to study the phylogenetic relationships and evolutionary history of Eurasian and African Hyalomma species has been advocated (Rees et al. 2003; Sands et al. 2017; Roth et al. 2019). The main objective of this work is to evaluate mtDNA cytochrome oxidase subunit (COI) gene diversity in H. lusitanicum populations to examine the biogeographic and phylogenetic relationships and determine the coherence of evolutionary lineages in Andalusia. This gene has focused increasing attention when it is used as a standardized DNA region to diagnose and delimit species by DNA barcoding (Hebert et al. 2003; Roe and Sperling 2007; Zhang and Zhang 2014).
Material and methods
Samples and DNA extraction
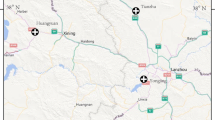
In total, 84 H. lusitanicum adult ticks were collected on vegetation by dragging in Andalusia, and 10 from other localities (Toledo, central Iberian Peninsula; Gran Canaria, Canary Island; and Sicily, Italy) (Fig. 1), mainly in summer, when adults of this species have the highest activity (Requena-García et al. 2017). After collection, the ticks were immediately placed in vials with 70% ethanol and properly labelled. Identification was confirmed in the laboratory based on taxonomic keys (Gil Collado et al. 1979; Manilla and Giannetto 1996; Apanaskevich et al. 2008).

Map of southern Spain, Canary Island and Sicily (Italy) indicating sampled localities and the haplotype frequency in each of the sites considered
Ticks were kept individually, rinsed with distilled water, dried on sterile filter paper and then crushed in sterile Eppendorf tubes. DNA was extracted using a DNA tissue kit according to the manufacturer’s instructions (Macherey–Nagel, Düren, Germany). During DNA extraction, we included sterile water as a negative control using a ratio of one control for every 15 samples. DNA extracts were stored at −20 °C until further processing.
Polymerase chain reaction (PCR)
A 887-bp fragment of the cytochrome oxidase subunit I (COI) gene was amplified and sequenced for each specimen. PCR amplifications were carried out in a MJ Mini Personal Thermal Cycler (Biorad, Hercules, CA, USA) using the primers hylCOIfor 5′-AATTTACAGTTTATCGCCT and HylCOIrev 5′-CATACAATAAAGCCTAATA (MWG, Ebersberg, Germany). Each PCR mixture consisted of the following: 4 μL of DNA, 20 pmol of each primer, 200 µM of dATP, dCTP, dTTP, dGTP, 2.0 mM MgCl2, 0.033 U of Biotaq DNA polymerase in 1 × PCR ammonium buffer (Bioline, Randolph, MA, USA), and sterile distilled water to a final volume of 80 μL. PCR cycles included an initial 90 s denaturation step at 96 °C, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 48 °C for 30 s, and extension at 72 °C for 50 s. Amplification was completed by holding the reaction mixture at 70 °C for 7 min to allow complete extension. PCR products were resolved by electrophoresis in 1.5% SeaKem agarose (Cambrex, Rockland, ME, USA) in 1 × buffer Bionic gels (Sigma, St. Louis, MO, USA) using a 100 bp ladder as molecular weight marker (Eurogentec, Seraing, Belgium). Products containing positive results were purified by using the Montage PCR kit (Millipore, Bedford, MA, USA) prior to sequencing. Negative controls were further processed by PCR as tick specimens. In order to reduce the risk of contamination no positive controls were used.
For the purpose of checking for potential amplification of nuclear mitochondrial paralogs (numts) (Moulton et al. 2010; Calvignac et al. 2011), we used two in silico strategies. First, the primers were aligned with the reference genome of Drosophila melanogaster using the UCSC (https://genome.ucsc.edu/cgi-bin/hgBlat)—both the forward and reverse primers did not display homology with any nuclear sequence. Second, the software Blast Primer (https://www.ncbi.nlm.nih.gov/tools/primer-blast) did not predict any amplification from the Ixodes scapularis nuclear genome.
Sequencing and sequence analysis
Positive PCR products were sequenced using PCR primers and the GenomeLab DTCS-Quick Start kit (Beckman Coulter, Fullerton, CA, USA) and a CEQ 2000XL capillary DNA sequencer (Beckman Coulter) according to the manufacturer’s instructions. In addition to forward and reverse primers two internal primers (HylCOIfor2 5′-GGATAACAATAGAACGTATAC and HylCOIrev2 5′-CAGTTCCTGCTCCTGATC) were used to read terminal regions. The forward and reverse sequences obtained were assembled into contigs with the program Bioedit v.7.0.1 (Hall 1999). The resulting COI sequences were manually aligned and analysed to obtain consensus sequences and to align and compare with other tick sequences found on GenBank database, using the BLAST feature (http://ncbi.nlm.nih.gov/blastn) (Altschul et al. 1990).
Phylogenetic analyses
The COI gene region was nearly length invariant for all the samples, making alignment trivial. Nucleotide variation and substitution patterns were examined using the software package MEGA7 (www.megasoftware.net; Kumar et al. 2016). Standard genetic indices, haplotype diversity (h) and nucleotide diversity (π), were computed.
Phylogenetic relationships between haplotypes were inferred by constructing webs connecting haplotypes of H. lusitanicum using the complete dataset using Median-joining and Templeton–Crandall–Sing (TCS) networks algorithms (Templeton et al. 1992; Clement et al. 2000), available in PopArt (http://popart.otago.ac.nz) (Bandelt et al. 1999). This program determines the number of polymorphic sites, h, π and the average number of nucleotide differences.
Molecular diversity indices were generated for seven populations and each gene using Arlequin (Excoffier and Lischer 2010). The population genetic structure and neutrality of populations were tested by analysis of molecular variance (AMOVA), including the overall fixation index statistics (FST) and pairwise FST with 1023 permutations.
Results
Nucleotide sequence of the COI region was determined for all 94 H. lusitanicum samples. Mitochondrial DNA polymorphism in the fragments showed 27 polymorphic sites that differentiated 17 distinct haplotypes (Fig. 1; GenBank accession codes and localities are presented in Table 1). The observed nucleotide frequencies were 0.295 (A), 0.385 (T/U), 0.173 (C), and 0.146 (G). The number of amino acid substitutions per site from mean diversity calculations for the entire population was 0.017.
For our dataset, MEGA7 suggested the T92+G model (Tamura 3-parameters; Tamura and Nei 1993). Given that the trees were best resolved, the maximum likelihood (ML) and neighbor joining (NJ) analysis were based on this model (value of –lnL = –1382.581; BIC = 3092.278; AICc = 2833.320 and gamma shape = 0.43).
The parsimony analysis between haplotypes indicated the existence of 860 constant characters. Among the 27 variable characters, seven were parsimony-informative and 20 were singleton sites. The unweighted parsimony search resulted in one phylogenetic tree. Maximum parsimony of the unweighted data yielded a tree length of 28 steps along with corresponding bootstrap percentages. The consistency index (CI) for the tree was 0.875 and the retention index (RI) 0.952. The frequencies obtained for each haplotype and the population of origin are represented in Fig. 2, the minimum spanning network is shown in Fig. 3. Nucleotide diversity (π) was 0.004, with seven parsimony-informative sites. Tajima D statistic has a calculated value of −0.963, whereas p (D ≤ −0.963) was 0.824.

COI haplotype frequencies for the seven sites considered in this study. The Andalusian sites are Cádiz, Córdoba, Huelva and Jaén

Nested clade design in the minimum spanning tree for Hyalomma lusitanicum mitochondrial haplotypes. Circle size is proportional to the number of similar haplotypes observed in the dataset. Small white circles indicate missing haplotypes. The number of bars between two circles represents the number of mutations between haplotypes
Because of unequal sample sizes, it is not always possible to compare the genetic diversity obtained from distinct geographical regions. Higher genetic variability is expected in source populations. Haplotypes 2 and 7 were present in four of the Iberian Peninsula sites plus in Canary Island (ht 2) and Sicily (ht 7) (Table 1). Seven haplotypes (3, 8, 9, 11, 12, 13 and 15) were found in only one site. In Andalusia, 16 of the 17 haplotypes identified in this study were found; only haplotype 12 appeared exclusively in a population outside the area (Toledo; Table 1, Fig. 2). Ten haplotypes appeared in the widely sampled and easternmost province of Jaén. Six other haplotypes detected in Andalusia were found in Jaén (Figs. 1 and 2).
The AMOVA analysis (Table 2) indicated that the COI gene exhibited high and significant differentiation among populations (FST = 0.053, P < 0.01). Most of the genetic variation (94.7%) was explained by differences among individuals within a population, whereas the remainder of the variation was attributable to differences among populations. Haplotype diversity (h) ranged from 0.720 (Toledo) to 0.860 (Córdoba), the mean value for the Iberian Peninsula populations was 0.815 (Table 3). Considering only Iberian ticks, 80% of the Shannon information index (sH) is due to differences within the five sites considered.
The graph representing the phylogenetic network visualizes population relationships defined by haplotypes separated by a maximum of 13 mutations (Fig. 3).
Discussion
Our mitochondrial data indicate low genetic diversity across the geographical range of H. lusitanicum in Andalusia. No genetic discontinuity is apparent between the tick populations, including specimens from Canary Island and Sicily.
Sands et al. (2017) show a phylogeny of Hyalomma ticks based on their evolutionary history in relation with tectonic events and large-scale environmental changes registered during a long period, with a divergence time around 10 million years ago. In this context, H. lusitanicum occupied a marginal area of distribution in the Iberian Peninsula and a scarce number of historically related island territories. The presence of this species out of this area is mainly anecdotic (Santos-Silva 2017). The low COI genetic diversity observed in Canary Islands and Sicily may be due to founder events and the maternal inheritance of mtDNA, but this hypothesis has to be confirmed using greater sample sizes. The results indicated that gene flow of the maternal lineage is relatively broad.
The distribution of H. lusitanicum haplotypes is the result of a long interaction of this species with its natural hosts. However, the current distribution of the various haplotypes has been influenced by anthropogenic action, manifested as habitat modification and the introduction of new host species through domestication (Hoberg and Brooks 2008; Ledger and Mitchell 2019). The movement of large masses of livestock (especially cattle, goats, sheep and horses) related to transhumance must have affected the phylogeography of the species in a relatively small area over time. This activity has been actively developed in the Iberian Peninsula over many centuries. Animal herders have used well-known travel routes, initially through the Roman roads and more recently through the glens, from the 13th century onwards (Walker 1983). This determined seasonal migrations of livestock from North to South and vice versa in a fall-spring cycle, concerning ovine, bovine, caprine and equine herds (Gómez Sal and Lorente 2004) that permit the gene flow among populations and haplotypes widespread in the distribution area of H. lusitanicum. Migratory movements also represent a unique dispersal mechanism (Bauer and Hoyer 2014), and long-distance dispersal events may be highly important for the re-colonization of unoccupied habitats, the recovery of lost populations and maintenance of gene flow (Viana et al. 2013). Bioclimatic conditions limit the actual distribution of Hyalomma species, as has been hypothesized by Estrada-Peña et al. (2011, 2012a, b) for H. marginatum. Wild fauna displacements, megafauna disappearance in southwest Europe (Malhi et al. 2016) and transhumance in recent times have contributed to the formation of a valuable set of sylvopastoral landscapes and associated natural habitats (Gómez Sal and Lorente 2004). Changes in the ecology of migratory species could have enormous impact on pathogen spread in wildlife and livestock, as well as altering human exposure to zoonotic infections (Hoberg et al. 2008; Altizer et al. 2011). Further investigation of the phylogeographic history of this species would be useful, increasing the sample size from various parts of its distribution area and increasing the number of genetic markers.
References
Almería S, Castellà J, Ferrer D, Ortuño A, Estrada-Peña A, Gutiérrez JF (2001) Bovine piroplasms in Minorca (Balearic Islands Spain): a comparison of PCR-based and light microscopy detection. Vet Parasitol 99:249–259. https://doi.org/10.1016/s0304-4017(01)00464-2
Altizer S, Bartel B, Han BA (2011) Animal migration and infectious disease risk. Science 331:296–302. https://doi.org/10.1126/science.1194694
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Apanaskevich DA (2004) Host-parasite relationships of the genus Hyalomma Koch 1844 (Acari Ixodidae) and their connection with microevolutionary process. Parazitologiia 38:515–523
Apanaskevich DA, Santos-Silva MM, Horak IG (2008) The genus Hyalomma Koch 1844. IV. Redescription of all parasitic stages of H. (Euhyalomma) lusitanicum Koch 1844 and the adults of H. (E.) franchinii Tonelli Rondelli 1932 (acari: ixodidae) with a first description of its immature stages. Folia Parasitol (Praha) 55:61–74. https://doi.org/10.14411/fp.2008.009
Avise JC (2004) Molecular markers natural history and evolution, 2nd edn. Sinauer Associates, Sunderland
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bauer S, Hoyer BJ (2014) Migratory animals couple biodiversity and ecosystem functioning worldwide. Science 344(6179):1242552. https://doi.org/10.1126/science.1242552
Brígido C, da Fonseca IP, Parreira R, Fazendeiro I, do Rosário VE, Centeno-Lima S, (2004) Molecular and phylogenetic characterization of Theileria spp. parasites in autochthonous bovines (Mirandesa breed) in Portugal. Vet Parasitol 123:17–23. https://doi.org/10.1016/j.vetpar.2004.05.024
Calvignac S, Konecny L, Malard F, Douady CJ (2011) Preventing the pollution of mitochondrial datasets with nuclear mitochondrial paralogs (numts). Mitochondrion 11:246–254. https://doi.org/10.1016/j.mito.2010.10.004
Caporale DA, Rich SM, Spielman A, Telford SR 3rd, Kocher TD (1995) Discriminating between Ixodes ticks by means of mitochondrial DNA sequences. Mol Phylogenet Evol 4:361–365. https://doi.org/10.1006/mpev.1995.1033
Castellà J, Estrada-Peña A, Almería S, Ferrer D, Gutiérrez J, Ortuño A (2001) A survey of ticks (Acari: Ixodidae) on dairy cattle on the island of Menorca in Spain. Exp Appl Acarol 25:899–908. https://doi.org/10.1023/a:1020482017140
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659. https://doi.org/10.1046/j.1365-294x.2000.01020.x
Estrada-Peña A, Santos-Silva MM (2005) The distribution of ticks (Acari: Ixodidae) of domestic livestock in Portugal. Exp Appl Acarol 36:233–246. https://doi.org/10.1007/s10493-005-5107-9
Estrada-Peña A, Quíez J, Sánchez Acedo C (2004) Species composition distribution and ecological preferences of the ticks of grazing sheep in north-central Spain. Med Vet Entomol 18:123–133. https://doi.org/10.1111/j.0269-283X.2004.00486.x
Estrada-Peña A, Martínez Avilés M, Muñoz Reoyo MJ (2011) A population model to describe the distribution and seasonal dynamics of the tick Hyalomma marginatum in the Mediterranean Basin. Transbound Emerg Dis 58:213–223. https://doi.org/10.1111/j.1865-1682.2010.01198.x
Estrada-Peña A, Palomar AM, Santibáñez P, Sánchez N, Habela MA, Portillo A, Romero L, Oteo J (2012) Crimean-Congo hemorrhagic fever virus in ticks southwestern Europe 2010. Emerg Infect Dis 18:179–180. https://doi.org/10.3201/eid1801.111040
Estrada-Peña A, Sánchez N, Estrada-Sánchez A (2012b) An assessment of the distribution and spread of the tick Hyalomma marginatum in the western Palearctic under different climate scenarios. Vector Borne Zoonotic Dis 12:758–768. https://doi.org/10.1089/vbz.2011.0771
Excoffier L, Lischer H (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
García-Sanmartín J, Nagore D, García-Pérez AL, Juste RA, Hurtado A (2006) Molecular diagnosis of Theileria and Babesia species infecting cattle in Northern Spain using reverse line blot macroarrays. BMC Vet Res 2:16. https://doi.org/10.1186/1746-6148-2-16
Georges K, Loria GR, Riili S, Greco A, Caracappa S, Jongejan F, Sparagano O (2001) Detection of haemoparasites in cattle by reverse line blot hybridisation with a note on the distribution of ticks in Sicily. Vet Parasitol 99:273–286. https://doi.org/10.1016/s0304-4017(01)00488-5
Gil Collado J, Guillen Llera JL, Zapatero Ramos LM (1979) Claves para la identificación de los Ixodidea españoles (adultos). Rev Iber Parasitol 39:107–111
Gomes J, Soares R, Santos M, Santos-Gomes G, Botelho A, Amaro A, Inácio J (2013) Detection of Theileria and Babesia infections amongst asymptomatic cattle in Portugal. Ticks Tick Borne Dis 4:148–151. https://doi.org/10.1016/j.ttbdis.2012.07.002
Gómez Sal A, Lorente I (2004) The Present status and ecological consequences of transhumance in Spain. In: Bunce RGH, Pérez-Soba M, Jongman RHG, Gómez Sal A, Herzog F, Austad I. (eds). Transhumance and biodiversity in European mountains Report of the EU-FP5 project TRANSHUMOUNT (EVK2-CT-2002-80017). IALE publication series nr 1, pp 233–248
González J, Valcárcel F, Pérez-Sánchez JL, Tercero-Jaime JM, Olmeda AS (2016) Seasonal dynamics of ixodid ticks on wild rabbits Oryctolagus cuniculus (Leporidae) from Central Spain. Exp Appl Acarol 70:369–380. https://doi.org/10.1007/s10493-016-0069-7
Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG (2014) The hard ticks of the World: (Acari: Ixodida: Ixodidae), 1st edn. Springer, Berlin
Habela M, Rol JA, Antón JM, Peña J, Corchero E, van Ham I, Jongejan E (1999) Epidemiology of Mediterranean theileriosis in Extremadura region Spain. Parassitologia 41:47–51
Hall TA (1999) Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hebert PD, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc Biol Sci 270:313–321. https://doi.org/10.1098/rspb.2002.2218
Hoberg EP, Brooks D (2008) A macroevolutionary mosaic: episodic host-switching, geographical colonization and diversification in complex host–parasite systems. J Biogeogr 35:1533–1550. https://doi.org/10.1111/j.1365-2699.2008.01951.x
Hoberg EP, Polley L, Jenkins EM, Kutz SJ (2008) Pathogens of domestic and free-ranging ungulates: global climate change in temperate to boreal latitudes across North America. Rev Sci Tech 27:511–528. https://doi.org/10.20506/rst.27.2.1818
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Ledger ML, Mitchell PD (2019) Tracing zoonotic parasite infections throughout human evolution. Int J Osteoarchaeol 29:l1-12. https://doi.org/10.1002/oa.2786
Malhi Y, Doughty CE, Galetti M, Smith FA, Svenning JC, Terborgh JW (2016) Megafauna and ecosystem function from the Pleistocene to the Anthropocene. Proc Nat Acad Sci USA 113:838–846. https://doi.org/10.1073/pnas.1502540113
Manilla G, Giannetto S (1996) Scanning electron microscopy study for an analytical key to the Hyalomminae (Ixodidae) species to be found in Italy. Parassitologia 38:537–541
Moraga-Fernández A, Ruiz-Fons F, Habela MA, Royo-Hernández L, Calero-Bernal R, Gortazar C, de la Fuente J, Fernández de Mera IG (2020) Detection of new Crimean-Congo haemorrhagic fever virus genotypes in ticks feeding on deer and wild boar. Transbound Emerg Dis Early view, Spain. https://doi.org/10.1111/tbed.13756
Moulton MJ, Song H, Whiting MF (2010) Assessing the effects of primer specificity on eliminating numt coamplification in DNA barcoding: a case study from Orthoptera (Arthropoda: Insecta). Mol Ecol Resour 10:615–627. https://doi.org/10.1111/j.1755-0998.2009.02823.x
Mtambo J, Madder M, Van Bortel W, Geysen D, Berkvens D, Backeljau T (2007) Genetic variation in Rhipicephalus appendiculatus (Acari: Ixodidae) from Zambia: correlating genetic and ecological variation with Rhipicephalus appendiculatus from eastern and southern Africa. J Vector Ecol 32:168–175. https://doi.org/10.3376/1081-1710(2007)32[168:gviraa]2.0.co;2
Murrell A, Campbell NJ, Barker SC (2001) A total-evidence phylogeny of ticks provides insights into the evolution of life cycles and biogeography. Mol Phylogenet Evol 21:244–258. https://doi.org/10.1006/mpev.2001.1018
Negredo A, Habela MÁ, de Arellano ER, Diez F, Lasala F, López P, Sarriá A, Labiod N, Calero-Bernal R, Arenas M, Tenorio A, Estrada-Peña A, Sánchez-Seco MP (2019) Survey of Crimean-Congo Hemorrhagic Fever Enzootic Focus Spain 2011–2015. Emerg Infect Dis 25:1177–1184. https://doi.org/10.3201/eid2506.180877
Ouhelli H (1994) Comparative development of Hyalomma marginatum (Koch 1844) H. detritum (Schuize 1919) H. anatolicum excavatum (Koch 1844) H. lusitanicum (Koch 1884) and H. dromedarii (Koch 1844) under laboratory conditions. Acta Parasitol 39:153–157
Ouhelli H, Pandey VS (1984) Development of Hyalomma lusitanicum under laboratory conditions. Vet Parasitol 15:57–66
Palomar AM, Portillo A, Santibáñez P, Mazuelas D, Arizaga J, Crespo A, Oteo JA (2013) Crimean-Congo hemorrhagic fever virus in ticks from migratory birds, Morocco. Emerg Infect Dis 19:260–263. https://doi.org/10.3201/eid1902.121193
Pereira A, Parreira R, Nunes M, Casadinho A, Vieira ML, Campino L, Maia C (2016) Molecular detection of tick-borne bacteria and protozoa in cervids and wild boars from Portugal. Parasit Vectors 9:251. https://doi.org/10.1186/s13071-016-1535-0
Rees DJ, Dioli M, Kirkendall LR (2003) Molecules and morphology: evidence for cryptic hybridization in African Hyalomma (Acari: Ixodidae). Mol Phylogenet Evol 27:131–142. https://doi.org/10.1016/s1055-7903(02)00374-3
Requena-García F, Cabrero-Sañudo F, Olmeda-García AS, González J, Valcárcel F (2017) Influence of environmental temperature and humidity on questing ticks in central Spain. Exp Appl Acarol 71:277–290. https://doi.org/10.1007/s10493-017-0117-y
Roe AD, Sperling FA (2007) Patterns of evolution of mitochondrial cytochrome c oxidase I and II DNA and implications for DNA barcoding. Mol Phylogenet Evol 44:325–345. https://doi.org/10.1016/j.ympev.2006.12.005
Roth A, Akad F, Zonstein I, King R, Orshan L, Erster O (2019) Molecular characterization of six Hyalomma species using mitochondrial markers. Ticks Tick Borne Dis 10:911–917. https://doi.org/10.1016/j.ttbdis.2019.04.015
Ruiz-Fons F, Fernández-de-Mera IG, Acevedo P, Höfle U, Vicente J, de la Fuente J, Gortazár C (2006) Ixodid ticks parasitizing Iberian red deer (Cervus elaphus hispanicus) and European wild boar (Sus scrofa) from Spain: geographical and temporal distribution. Vet Parasitol 140:133–142. https://doi.org/10.1016/j.vetpar.2006.03.033
Sands AF, Apanaskevich DA, Matthee S, Horak IG, Harrison A, Karim S, Mohammad MK, Mumcouglu KY, Rajakaruna RS, Santos-Silva M, Matthee CA (2017) Effects of tectonics and large scale climatic changes on the evolutionary history of Hyalomma ticks. Mol Phylogenet Evol 114:153–165. https://doi.org/10.1016/j.ympev.2017.06.002
Santos-Silva MM (2017) Hyalomma lusitanicum Koch 1844 (Figs. 155–157). In: Estrada-Peña A, Mihalca A, Petney T (eds) Ticks of Europe and North Africa. Springer, Cham, pp 383–387
Santos-Silva MM, Beati L, Santos AS, De Sousa R, Núncio MS, Melo P, Santos-Reis M, Fonseca C, Formosinho P, Vilela C, Bacellar F (2011) The hard-tick fauna of mainland Portugal (Acari: Ixodidae): an update on geographical distribution and known associations with hosts and pathogens. Exp Appl Acarol 55:85–121. https://doi.org/10.1007/s10493-011-9440-x
Sayin F, Karaer Z, Dincer S, Cakmak A, Inci A, Yukari BA, Eren H, Vatansever Z, Nalbantoglu S, Melrose TR (2003) A comparison of susceptibilities to infection of four species of Hyalomma ticks with Theileria annulata. Vet Parasitol 113:115–121. https://doi.org/10.1016/s0304-4017(03)00045-1
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease map** and DNA sequence data III Cladogram Estimation. Genetics 132:619–633
Torina A, Khoury C, Caracappa S, Maroli M (2006) Ticks infesting livestock on farms in Western Sicily Italy. Exp Appl Acarol 38:75–86. https://doi.org/10.1007/s10493-005-5629-1
Torina A, Alongi A, Scimeca S, Vicente J, Caracappa S, de la Fuente J (2010) Prevalence of tick-borne pathogens in ticks in Sicily. Transbound Emerg Dis 57:46–48. https://doi.org/10.1111/j.1865-1682.2010.01101.x
Valcárcel F, González J, Sánchez JP, Jaime JT, Olmeda AS (2016) Long-term ecological study of host-seeking adults of Hyalomma lusitanicum (Acari: Ixodidae) in a Meso-Mediterranean climate. J Med Entomol 53:221–224. https://doi.org/10.1093/jme/tjv152
Viana DS, Santamaría L, Michot TC, Figuerola J (2013) Migratory strategies of waterbirds shape the continental-scale dispersal of aquatic organisms. Ecography 36:430–438. https://doi.org/10.1111/j.1600-0587.2012.07588.x
Viseras J, Hueli LE, Adroher FJ, García-Fernández P (1999) Studies on the transmission of Theileria annulata to cattle by the tick Hyalomma lusitanicum. Zentralbl Veterinarmed B 46:505–509. https://doi.org/10.1111/j.1439-0450.1999.tb01242.x
Walker MJ (1983) Laying a mega-myth: Dolmens and drovers in prehistoric Spain. World Archaeol 15:37–50
Zhang RL, Zhang B (2014) Prospects of using DNA barcoding for species identification and evaluation of the accuracy of sequence databases for ticks (Acari: Ixodida). Ticks Tick Borne Dis 5:352–358. https://doi.org/10.1016/j.ttbdis.2014.01.001
Acknowledgements
We greatly appreciate the support of those who helped collect many of the specimens used in this study. The research activities of the authors are partially funded by the PAIDI (Junta de Andalucía): BIO-294 group. Technical and human support provided by Centro de Instrumentación Científico-Técnica (CICT)—Servicios Centrales de Apoyo a la Investigación (SCAI) of Universidad de Jaén (MICINN, Junta de Andalucía, FEDER) is gratefully acknowledged. We thank two anonymous referees for valuable comments on the manuscript.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Márquez, F.J., Caruz, A. Phylogeography of Hyalomma (Euhyalomma) lusitanicum (Acarina, Parasitiformes, Ixodidae) in Andalusia based on mitochondrial cytochrome oxidase I gene. Exp Appl Acarol 85, 49–61 (2021). https://doi.org/10.1007/s10493-021-00652-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-021-00652-0