
Abstract
Self-limiting focal epilepsies are among the most common forms of epilepsy in children. Based on family studies, a genetic basis is assumed for the epilepsy as well as the typical electroencephalographic (EEG) feature of centrotemporal spikes, although complex inheritance and possibly additional influencing factors must be considered. Variants in GRIN2A, encoding the GluN2A subunit of the N‑methyl-D-aspartate (NMDA) glutamate receptor, represent the most important genetic risk factor to date. With memantine for variants with a gain-of-function effect and L‑serine for loss-of-function variants, two personalized therapeutic approaches are potentially available. Their effectiveness and significance need to be clarified in further investigations and clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Self-limited focal epilepsy of childhood—formerly known as idiopathic focal epilepsy—comprises a group of disorders that occur in an age-related manner, have common characteristics, but can differ significantly in terms of manifestation, severity, and prognosis. The genetic causes of these forms of epilepsy have been discussed for many years, but their decoding and the translation of scientific findings into everyday clinical practice are still far less advanced than the frequency of the clinical picture would suggest.
In recent years, research has made steady progress in deciphering the genetic causes of epilepsy. In particular, knowledge of the genes responsible for epilepsy and developmental and epileptic encephalopathy (DEE), which usually begin early and are severe, is growing almost daily. At the same time, diagnostic genetic testing in affected children and adolescents is increasingly becoming part of clinical routine: Early after initial manifestation, comprehensive genetic tests are recommended and carried out to clarify the cause, whereby today, exome sequencing should be the method of first choice [3, 12]. The situation for self-limited focal epilepsy in childhood is different—the reasons for and consequences of this, as well as positive outlook, are presented here.
Family history and genetic pedigrees
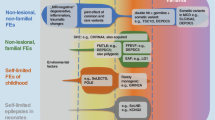
Familial clusters of focal childhood epilepsy have been described since the 1960s [4] and are certainly known to every practicing pediatric epileptologist. It is striking that the severity of the epilepsy can vary quite considerably, ranging from randomly discovered, asymptomatic electroencephalographic (EEG) changes with the occurrence of the typical, sleep-activated, often centrotemporal and comparatively well-structured spike-waves and sharp-slow waves (centrotemporal spikes = CTS) to rolandic epilepsy with few seizure events (new nomenclature: self-limited epilepsy with centrotemporal spikes, SeLECTS) to more severe epilepsy with multiple seizure forms and/or bioelectric status during sleep (previously: pseudo-Lennox syndrome or atypical benign partial epilepsy and electrical status epilepticus in sleep [ESES]/continuous spikes in slow wave [CSWS], summarized in the new classification of syndromes as epileptic encephalopathy with spike-wave activation in sleep). The age-dependent onset of symptoms, combined with the “disappearance” of typical EEG findings and seizures during puberty, makes genetic testing in families more difficult, meaning that these may no longer be detectable in later diagnostic testing, for example, for segregation analyses in families.
A recent meta-analysis of various (older) studies on the occurrence of isolated EEG changes in asymptomatic relatives/siblings of individuals with self-limited focal epilepsy shows EEG abnormalities (not exclusively CTS) in about 30% of the siblings studied [30]. Family studies for various forms of self-limited focal epilepsy show a positive family history for epilepsy in up to 60%, but the epilepsy in the family shows a wide phenotypic range and is not limited to epilepsy with CTS [8, 31, 33]. These results, together with twin studies showing a higher concordance for the EEG feature of centrotemporal spikes/sharp-slow-waves (CTS) than for the form of epilepsy [32] and family studies showing an autosomal dominant inheritance for the typical EEG abnormalities [1], thus indicate complex inheritance.
Beyond the spectrum of tuberous sclerosis: genes of the mTOR signaling pathway
In addition to the TSC1 and TSC2 genes, which are well-known in the context of tuberous sclerosis and whose products act as regulators of the mTOR signaling pathway, other genes/proteins in this signaling pathway that are relevant for developmental disorders, brain malformations, and epilepsy have been identified in recent years [34]. For example, pathogenic variants in DEPDC5 have been found in individuals with various forms of focal epilepsy, particularly in families with focal epilepsy with variable foci. Lal and colleagues studied a cohort of 207 patients with rolandic epilepsy and identified three pathogenic variants (= 1.4%); they also found variants in DEPDC5 in three of 82 families (= 3.7%) with unclassified, non-lesional focal childhood epilepsy in all affected individuals [16]. Further confirmatory studies have not yet been published, nor have any changes been identified in the other genes of the GATOR1 complex. Epilepsy that is characterized by genetically determined overactivity of the mTOR pathway could be amenable to medication with mTOR inhibitors such as rapamycin or everolimus, although this has not yet been investigated in studies, with the exception of epilepsy in tuberous sclerosis.
Infobox 1
Targeted testing of the genes of the mTOR signaling pathway, in particular the GATOR1 complex (DEPDC5, NPRL2, and NPRL3), e.g., using a gene panel or exome sequencing, is currently not useful for patients with SeLECTS. However, in familial and sporadic focal epilepsy in childhood that does not belong to the rolandic spectrum, targeted diagnostic testing to clarify the cause, genetic counseling, and, if necessary, the future use of precision treatment using mTOR inhibitors is recommended at an early stage [3, 12]. Diagnostic genetic testing may also be useful in lesional epilepsy prior to epilepsy surgery to better assess the risk of recurrence.
Microdeletions and microduplications
Microdeletions and microduplications can cause complex syndromic diseases as recurrent events (e.g., deletion 15q11q13 as the cause of Angelman syndrome) or, in the form of copy number variations (CNV), increase the risk of develo** common diseases (e.g., deletion 15q13.3 as a risk factor for generalized epilepsy). While the array comparative genomic hybridization (CGH) technique primarily used to date could only detect larger deletions and duplications (depending on the resolution of the array, i.e., the number of oligonucleotides or single nucleotide polymorphisms, SNPs, examined), data from exome sequencing using next-generation sequencing technology is now also used for CNV analysis. This also enables the detection of smaller, intragenic deletions and duplications that comprise, for example, only a few exons.
In 2010, Reutlinger and colleagues described three patients with overlap** microdeletions in the 16p13 chromosomal region [25]. The index patient had epilepsy classified as pseudo-Lennox syndrome, another patient had unclassified epilepsy with an ESES-like EEG pattern, and the third patient had epilepsy with similarities to rolandic epilepsy. All three also showed developmental disorders and facial dysmorphia. GRIN2A, coding for the alpha‑2 subunit (Glu2A) of the N-methyl-D-aspartate (NMDA) receptor, was identified as a common candidate gene for epilepsy.
There are no other known recurrent microdeletion syndromes that show the typical picture of SeLECTS as a hallmark symptom. Rare CNVs have also been identified in various studies in individuals with rolandic epilepsy, but appear to be rarer in this patient group than, for example, in genetically generalized forms of epilepsy [7, 11].
Infobox 2
CNV analysis is particularly useful in the clinical setting when epilepsy with aspects of SeLECTS or epileptic encephalopathy with spike-wave activation in sleep occurs as part of a syndromic clinical picture [3, 12]. CNV analysis from exome sequencing data is recommended, as it is more sensitive and economical than the previously widely used array CGH analysis.
The spectrum of epilepsy and developmental speech and language disorders: the significance of GRIN2A
Focal childhood epilepsy often shows a mild course, but can be accompanied by speech development disorders of varying severity. Scheffer and colleagues coined the term “epilepsy-aphasia spectrum disorder” to describe the severest manifestation of epilepsy with accompanying acoustic agnosia and subsequent aphasia consistent with Landau–Kleffner syndrome [31].
After identifying patients with microdeletion 16p13.2 (see above, [25]) and a few patients with developmental disorders, epilepsy, and mutations in the GRIN2A and GRIN2B genes [9], the most likely candidate gene for the expression of epilepsy, GRIN2A, was investigated in larger cohorts of patients with childhood focal epilepsy. NMDA glutamate receptors are ligand-gated ion channels formed by two glycine-binding GluN1 subunits (encoded by GRIN1) and two glutamate-binding GluN2 subunits (encoded by GRIN2A‑D). The composition of the GluN2 subunits varies depending on localization and age, with GluN2A (encoded by GRIN2A) becoming increasingly important after birth and in the first years of life [28].
At the same time, three research groups were able to elucidate and publish the significance of this gene for focal epilepsy, especially epilepsy with developmental speech and language disorders [5, 17, 18]. Further studies have confirmed these results with similar frequencies of pathogenic variants in smaller cohorts [6, 7, 19, 21]. These variants were found in patients with all known variants of self-limited focal epilepsy and were also identified in family members who only showed the EEG feature of CTS, but all of the studies describe a clear tendency toward more frequent findings of mutation in the more severe clinical pictures (“atypical Rolandic epilepsy”, Landau–Kleffner syndrome, and ESES/epileptic encephalopathy with spike-wave activation in sleep). As an example, by Lemke and colleagues [17], variants were found in 27 of 359 patients (= 7.5%) with self-limited focal epilepsy, whereby 12 of 245 (= 4.9%) individuals with SeLECTS (rolandic epilepsy/benign epilepsy with centrotemporal spikes [BECTS]) and 9 of 51 (= 17.6%) individuals with epileptic encephalopathy with spike-wave activation in sleep (CSWS) showed pathogenic variants in GRIN2A.
Infobox 3
A comprehensive collection of variants and associated phenotypes for both GRIN2A and other GRIN-related disorders is being compiled in the GRIN Registry, curated by the Institute of Human Genetics at Leipzig University (Prof. Johannes Lemke, Dr. Ilona Krey) together with working groups in Denver, Atlanta, and Cleveland, USA (http://www.redcap.medizin.uni-leipzig.de/redcap/surveys/?s=PRAEF9N7J7). The associated GRIN portal can be accessed via www.grin-portal.broadinstitute.org and offers scientists, practitioners, and affected individuals comprehensive information on GRIN-related disorders and their genetic basis. The Leipzig Institute of Human Genetics also offers specialized consultation hours, which can be accessed via https://www.uniklinikum-leipzig.de/einrichtungen/humangenetik/genetische-sprechstunde. The German family support group Gemeinsam GRIN (“GRIN Together”) brings together families affected by various GRIN-related clinical pictures, runs a closed Facebook group, and can be contacted by email at Gemeinsam-grin@gmx.de.

There are also international, English-speaking family support groups that should also be mentioned here: CureGRIN Foundation (www.curegrin.org) and Grin Europe (www.grineurope.org). Functional studies of different variants show both gain-of-function (GOF) effects and loss-of-function (LOF). The extensive work of Strehlow and colleagues [28] describes genotype–phenotype correlations between variant type and clinical presentation (missense variants in transmembrane and linker domains of the protein are associated with severe phenotypes related to epilepsy, developmental disorders, and other abnormalities, while missense variants of the amino end and ligand-binding domains cause milder disease). This correlation is reflected in functional data from electrophysiological studies: Here, the variants are often responsible for a GOF effect in severe disease, while the milder diseases appear to be caused by LOF effects. It has been widely discussed that the variants influence the physiological change in expression and functionality from GluN2B (GRIN2B) to GluN2A (GRIN2A) during brain development. Data from the aforementioned study by Strehlow and colleagues, which also investigated the expression of GluN2B as well as GluN2B-dependent currents in a GRIN2A knock-out rat model, contradict this assumption, as no compensation by GluN2B was detected either in gene expression or in electrophysiology [28].
The ultimate goal of all genetic research and diagnostics is the identification of pathogenetically relevant changes that allow for improved and targeted treatment in the sense of precision medicine. For GRIN2A-related epilepsy with GOF mutations, great hopes were therefore pinned on treatment with the NMDA receptor antagonist memantine, after an initial case report described clearly positive effects of this drug [22]. Unfortunately, confirmatory studies provide only limited support for therapeutic decisions, which means that the treatment success of individual cases is of little significance. Disease patterns associated with LOF variants are potentially amenable to treatment with L‑serine, which has an agonistic effect on the NMDA receptor via its enantiomer, D‑serine. After positive effects were initially described by Sotos and colleagues in an individual with a pathogenic GRIN2B variant and severe encephalopathy [27], a recently published case series reported improvements (to varying degrees) in epilepsy, EEG, cognition, and behavior in nine additional patients, three of eight had an LOF variant in GRIN2A, while six had variants in GRIN2B [13]. Interestingly, one girl with a GOF variant showed an immediate worsening of symptoms after the erroneous initiation of L‑serine treatment [13]. Further studies need to differentiate and confirm the effects of this therapeutic approach with regard to epilepsy and cognition, and further data on dose finding and tolerability need to be collected.
Infobox 4
GRIN2A is one of the important differential diagnoses in cases of severe disease with epilepsy and/or speech development disorders (with or without an abnormal family history) and can be most efficiently investigated in the context of exome diagnostics [3, 12].
It is important for the evaluation of test results that even abnormal findings consistent with a genetic risk factor probably only explain partial aspects of the cause of the disease and pathophysiology. For patients with LOF variants, there are initial indications that L‑serine could provide a simple, well-tolerated, and potentially effective treatment.
Genetic findings of uncertain significance
The first genetic studies were carried out in the 1990s in the form of linkage analyses to narrow down the relevant chromosomal regions. For SeLECTS (then BECTS), linkage to the chromosomal region 15q14 was found [20]. The CHRNA7 gene, which codes for subunit 4 of the N-acetylcholine receptor, was seen as a candidate gene in this region. However, no causative changes in this gene have yet been identified in patients with SeLECTS. Linkage analysis for the EEG feature of CTS found a linkage to chromosomal region 11p13 with the candidate gene ELP4, coding for the elongator protein complex 4, which, as part of the Elongator complex, is involved in transcription and tRNA modification [29]. Further studies have not yet been able to confirm a connection between changes in ELP4 and CTS/rolandic epilepsy [24].
In 2013, Lal and colleagues found five of 289 patients with rolandic epilepsy (= 1.7%) to have intragenic deletions and variants in the RBFOX1 gene, a regulator of alternative mRNA splicing of various neuronal genes, that segregated in the corresponding families with the disease [15]. The same research group also investigated the presence of exon-encompassing deletions in a cohort of 807 individuals with sporadic focal epilepsy and identified alterations in 0.9% of patients [14]. These patients suffered from both MRI-negative focal epilepsy (n = 5) and structural focal epilepsy in the presence of hippocampal sclerosis (n = 2). Rarely, intragenic deletions were also found in individuals with generalized epilepsy and were interpreted as a genetic risk factor. As also shown for other genes, the phenotypic spectrum of RBFOX1-associated epilepsy appears to be correspondingly broad and not limited to “idiopathic” forms of epilepsy.
More recent studies have identified further candidate genes whose relevance to the clinical picture of self-limited focal epilepsy remains to be demonstrated [2, 10, 23]. In 2020, the working group of Lesca and colleagues published a study in which 57 child–parent trios were examined by exome sequencing, who initially tested negative for changes in GRIN2A (see below) [26]. 20 patients, the majority with rare and severe forms of focal epilepsy, showed alterations in potential candidate genes; individual variants in GRIN2B and CAMK2A were considered likely to be relevant. CAMK2A (calcium/calmodulin-dependent protein kinase II) had previously been identified as a relevant gene in cognitive developmental disorders with and without concomitant epilepsy (OMIM: MRD53, #617798), while GRIN2B is known as the gene responsible for epileptic encephalopathies with variable additional symptoms such as brain malformations, developmental disorders, autism, and movement disorders (OMIM: DEE27, #616139).
Practical conclusion
-
The genetics of self-limited focal epilepsy is complex: Various candidate genes have been identified that can be classified as genetic risk factors. Their evaluation in a clinical context is difficult.
-
Therefore, diagnostic genetic testing appears to be useful and advisable in severe forms of the disease, presence of a positive family history, and/or additional symptoms that suggest syndromic disorders/contiguous gene syndromes.
-
In more common forms of SeLECTS, the diagnostic and prognostic value of genetic testing is considered to be low.
-
There are initial hopes for personalized treatments following positive reports on the use of L‑serine in patients with mutations in the genes coding for N-methyl-D-aspartate (NMDA) receptor subunits GRIN2A and GRIN2B, for which a loss-of-function effect has been demonstrated.
References
Bali B, Kull LL, Strug LJ, Clarke T, Murphy PL, Akman CI, Greenberg DA, Pal DK (2007) Autosomal dominant inheritance of centrotemporal sharp waves in rolandic epilepsy families. Epilepsia 48:2266–2272
Bobbili DR, Lal D, May P, Reinthaler EM, Jabbari K, Thiele H, Nothnagel M, Jurkowski W, Feucht M, Nürnberg P, Lerche H, Zimprich F, Krause R, Neubauer BA, Reinthaler EM, Zimprich F, Feucht M, Steinböck H, Neophytou B, Geldner J, Gruber-Sedlmayr U, Haberlandt E, Ronen GM, Altmüller J, Lal D, Nürnberg P, Sander T, Thiele H, Krause R, May P, Balling R, Lerche H, Neubauer BA (2018) Exome-wide analysis of mutational burden in patients with typical and atypical rolandic epilepsy. Eur J Hum Genet 26:258–264. https://doi.org/10.1038/s41431-017-0034-x
Boßelmann C, Borggräfe I, Fazeli W, Klein K‑M, Kluger GJ, Müller-Schlüter K, Neubauer BA, von Spiczak S, Steinbeis von Stülpnagel C, Weber Y, Lemke JR, Wolking S, Krey I (2023) Genetische Diagnostik der Epilepsien: Empfehlung der Kommission Epilepsie und Genetik der Deutschen Gesellschaft für Epileptologie (DGfE). Clin Epileptol 36:224–237. https://doi.org/10.1007/s10309-023-00580-6
Bray PF, Wiser WC (1965) Hereditary characteristics of familial temporal-central focal epilepsy. Pediatrics 36:207–211
Carvill GL, Regan BM, Yendle SC, O’Roak BJ, Lozovaya N, Bruneau N, Burnashev N, Khan A, Cook J, Geraghty E, Sadleir LG, Turner SJ, Tsai MH, Webster R, Ouvrier R, Damiano JA, Berkovic SF, Shendure J, Hildebrand MS, Szepetowski P, Scheffer IE, Mefford HC (2013) GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 45:1073–1076
DeVries SP, Patel AD (2014) Two patients with a GRIN2A mutation and childhood-onset epilepsy. Pediatr Neurol 49:482–485
Dimassi S, Labalme A, Lesca G, Rudolf G, Bruneau N, Hirsch E, Arzimanoglou A, Motte J, de Martin Saint A, Boutry-Kryza N, Cloarec R, Benitto A, Ameil A, Edery P, Ryvlin P, De Bellescize J, Szepetowski P, Sanlaville D (2014) A subset of genomic alterations detected in rolandic epilepsies contains candidate or known epilepsy genes including GRIN2A and PRRT2. Epilepsia 55:370–378
Doose H, Neubauer BA, Petersen B (2000) The concept of hereditary impairment of brain maturation. Epileptic Disord Int Epilepsy J Videotape 2(1):S45–S49
Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortum F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, Moog U, Rappold G, Rauch A, Ropers HH, von Spiczak S, Tonnies H, Villeneuve N, Villard L, Zabel B, Zenker M, Laube B, Reis A, Wieczorek D, Van Maldergem L, Kutsche K (2010) Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42:1021–1026
Hu X, Tang J, Hua Y, Wang Y, Huang J (2021) Evaluation of candidate genes in a Chinese cohort of atypical Rolandic epilepsy. Epileptic Disord Int Epilepsy J Videotape 23:623–632. https://doi.org/10.1684/epd.2021.1308
Jabbari K, Bobbili DR, Lal D, Reinthaler EM, Schubert J, Wolking S, Sinha V, Motameny S, Thiele H, Kawalia A, Altmüller J, Toliat MR, Kraaij R, van Rooij J, Uitterlinden AG, Ikram MA, Zara F, Lehesjoki A‑E, Krause R, Zimprich F, Sander T, Neubauer BA, May P, Lerche H, Nürnberg P (2018) Rare gene deletions in genetic generalized and rolandic epilepsies. PLoS ONE 13:e202022. https://doi.org/10.1371/journal.pone.0202022
Krey I, Platzer K, Lemke JR (2022) Monogenetic epilepsies and how to approach them in 2022. Med Genet 34:201–205. https://doi.org/10.1515/medgen-2022-2143
Krey I, von Spiczak S, Johannesen KM, Hikel C, Kurlemann G, Muhle H, Beysen D, Dietel T, Møller RS, Lemke JR, Syrbe S (2022) L‑Serine treatment is associated with improvements in behavior, EEG, and seizure frequency in individuals with GRIN-related disorders due to null variants. Neurother J Am Soc Exp Neurother 19:334–341. https://doi.org/10.1007/s13311-021-01173-9
Lal D, Pernhorst K, Klein KM, Reif P, Tozzi R, Toliat MR, Winterer G, Neubauer B, Nürnberg P, Rosenow F, Becker F, Lerche H, Kunz WS, Kurki MI, Hoffmann P, Becker AJ, Perucca E, Zara F, Sander T, Weber YG (2015) Extending the phenotypic spectrum of RBFOX1 deletions: sporadic focal epilepsy. Epilepsia 56:e129–133. https://doi.org/10.1111/epi.13076
Lal D, Reinthaler EM, Altmüller J, Toliat MR, Thiele H, Nürnberg P, Lerche H, Hahn A, Møller RS, Muhle H, Sander T, Zimprich F, Neubauer BA (2013) RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS ONE 8:e73323. https://doi.org/10.1371/journal.pone.0073323
Lal D, Reinthaler EM, Schubert J, Muhle H, Riesch E, Kluger G, Jabbari K, Kawalia A, Baumel C, Holthausen H, Hahn A, Feucht M, Neophytou B, Haberlandt E, Becker F, Altmuller J, Thiele H, Lemke JR, Lerche H, Nurnberg P, Sander T, Weber Y, Zimprich F, Neubauer BA (2014) DEPDC5 mutations in genetic focal epilepsies of childhood. Ann Neurol 75:788–792
Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, Geider K, Laube B, Schwake M, Finsterwalder K, Franke A, Schilhabel M, Jahn JA, Muhle H, Boor R, Van Paesschen W, Caraballo R, Fejerman N, Weckhuysen S, De Jonghe P, Larsen J, Moller RS, Hjalgrim H, Addis L, Tang S, Hughes E, Pal DK, Veri K, Vaher U, Talvik T, Dimova P, Guerrero Lopez R, Serratosa JM, Linnankivi T, Lehesjoki AE, Ruf S, Wolff M, Buerki S, Wohlrab G, Kroell J, Datta AN, Fiedler B, Kurlemann G, Kluger G, Hahn A, Haberlandt DE, Kutzer C, Sperner J, Becker F, Weber YG, Feucht M, Steinbock H, Neophythou B, Ronen GM, Gruber-Sedlmayr U, Geldner J, Harvey RJ, Hoffmann P, Herms S, Altmuller J, Toliat MR, Thiele H, Nurnberg P, Wilhelm C, Stephani U, Helbig I, Lerche H, Zimprich F, Neubauer BA, Biskup S, von Spiczak S (2013) Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 45:1067–1072
Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J, Wright S, Tsintsadze V, Michel A, Doummar D, Lascelles K, Strug L, Waters P, de Bellescize J, Vrielynck P, de Martin Saint A, Ville D, Ryvlin P, Arzimanoglou A, Hirsch E, Vincent A, Pal D, Burnashev N, Sanlaville D, Szepetowski P (2013) GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 45:1061–1066
Li X, **e L‑L, Han W, Hong S‑Q, Ma J‑N, Wang J, Jiang L (2020) Clinical forms and GRIN2A genotype of severe end of epileptic-aphasia spectrum disorder. Front Pediatr 8:574803. https://doi.org/10.3389/fped.2020.574803
Neubauer BA, Fiedler B, Himmelein B, Kampfer F, Lassker U, Schwabe G, Spanier I, Tams D, Bretscher C, Moldenhauer K, Kurlemann G, Weise S, Tedroff K, Eeg-Olofsson O, Wadelius C, Stephani U (1998) Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14. Neurology 51:1608–1612
Pavlidis E, Møller RS, Nikanorova M, Kölmel MS, Stendevad P, Beniczky S, Tassinari CA, Rubboli G, Gardella E (2019) Idiopathic encephalopathy related to status epilepticus during slow sleep (ESES) as a “pure” model of epileptic encephalopathy. An electroclinical, genetic, and follow-up study. Epilepsy Behav EB 97:244–252. https://doi.org/10.1016/j.yebeh.2019.05.030
Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, Golas G, Simeonov DR, Holloman C, Tankovic A, Karamchandani MM, Schreiber JM, Mullikin JC, Tifft CJ, Toro C, Boerkoel CF, Traynelis SF, Gahl WA (2014) GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol 1:190–198
Ramos LLP, Monteiro FP, Sampaio LPB, Costa LA, Ribeiro MDO, Freitas EL, Kitajima JP, Kok F (2019) Heterozygous loss of function of NR4A2 is associated with intellectual deficiency, rolandic epilepsy, and language impairment. Clin Case Rep 7:1582–1584. https://doi.org/10.1002/ccr3.2260
Reinthaler EM, Lal D, Jurkowski W, Feucht M, Steinböck H, Gruber-Sedlmayr U, Ronen GM, Geldner J, Haberlandt E, Neophytou B, Hahn A, Altmüller J, Thiele H, Toliat MR, EuroEPINOMICS Consortium, Lerche H, Nürnberg P, Sander T, Neubauer BA, Zimprich F (2014) Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 55:e89–e93. https://doi.org/10.1111/epi.12712
Reutlinger C, Helbig I, Gawelczyk B, Subero JI, Tonnies H, Muhle H, Finsterwalder K, Vermeer S, Pfundt R, Sperner J, Stefanova I, Gillessen-Kaesbach G, von Spiczak S, van Baalen A, Boor R, Siebert R, Stephani U, Caliebe A (2010) Deletions in 16p13 including GRIN2A in patients with intellectual disability, various dysmorphic features, and seizure disorders of the rolandic region. Epilepsia 51:1870–1873
Rudolf G, de Bellescize J, de Saint MA, Arzimanoglou A, Valenti Hirsch MP, Labalme A, Boulay C, Simonet T, Boland A, Deleuze JF, Nitschké P, Ollivier E, Sanlaville D, Hirsch E, Chelly J, Lesca G (2020) Exome sequencing in 57 patients with self-limited focal epilepsies of childhood with typical or atypical presentations suggests novel candidate genes. Eur J Paediatr Neurol Ejpn Off J Eur Paediatr Neurol Soc 27:104–110. https://doi.org/10.1016/j.ejpn.2020.05.003
Soto D, Olivella M, Grau C, Armstrong J, Alcon C, Gasull X, Santos-Gómez A, Locubiche S, Gómez de Salazar M, García-Díaz R, Gratacòs-Batlle E, Ramos-Vicente D, Chu-Van E, Colsch B, Fernández-Dueñas V, Ciruela F, Bayés À, Sindreu C, López-Sala A, García-Cazorla À, Altafaj X (2019) L‑Serine dietary supplementation is associated with clinical improvement of loss-of-function GRIN2B-related pediatric encephalopathy. Sci Signal 12:eaaw936. https://doi.org/10.1126/scisignal.aaw0936
Strehlow V, Heyne HO, Vlaskamp DRM, Marwick KFM, Rudolf G, de Bellescize J, Biskup S, Brilstra EH, Brouwer OF, Callenbach PMC, Hentschel J, Hirsch E, Kind PC, Mignot C, Platzer K, Rump P, Skehel PA, Wyllie DJA, Hardingham GE, van Ravenswaaij-Arts CMA, Lesca G, Lemke JR, GRIN2A study group (2019) GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain J Neurol 142:80–92. https://doi.org/10.1093/brain/awy304
Strug LJ, Clarke T, Chiang T, Chien M, Baskurt Z, Li W, Dorfman R, Bali B, Wirrell E, Kugler SL, Mandelbaum DE, Wolf SM, McGoldrick P, Hardison H, Novotny EJ, Ju J, Greenberg DA, Russo JJ, Pal DK (2009) Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4). Eur J Hum Genet 17:1171–1181
Tashkandi M, Baarma D, Tricco AC, Boelman C, Alkhater R, Minassian BA (2019) EEG of asymptomatic first-degree relatives of patients with juvenile myoclonic, childhood absence and rolandic epilepsy: a systematic review and meta-analysis. Epileptic Disord Int Epilepsy J Videotape 21:30–41. https://doi.org/10.1684/epd.2019.1024
Tsai M‑H, Vears DF, Turner SJ, Smith RL, Berkovic SF, Sadleir LG, Scheffer IE (2013) Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia 54:280–287. https://doi.org/10.1111/epi.12065
Vadlamudi L, Kjeldsen MJ, Corey LA, Solaas MH, Friis ML, Pellock JM, Nakken KO, Milne RL, Scheffer IE, Harvey AS, Hopper JL, Berkovic SF (2006) Analyzing the etiology of benign rolandic epilepsy: a multicenter twin collaboration. Epilepsia 47:550–555
Vears DF, Tsai M‑H, Sadleir LG, Grinton BE, Lillywhite LM, Carney PW, Simon Harvey A, Berkovic SF, Scheffer IE (2012) Clinical genetic studies in benign childhood epilepsy with centrotemporal spikes. Epilepsia 53:319–324. https://doi.org/10.1111/j.1528-1167.2011.03368.x
Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I, Baulac M, Fohlen M, Kallay Zetchi C, Seeck M, de la Grange P, Dermaut B, Meurs A, Thomas P, Chassoux F, Leguern E, Picard F, Baulac S (2016) Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 57:994–1003. https://doi.org/10.1111/epi.13391
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
I. Krey, J.R. Lemke and S. von Spiczak declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies mentioned were in accordance with the ethical standards as indicated in the respective publications.
The supplement containing this article is not sponsored by industry.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

Scan QR code & read article online
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krey, I., Lemke, J.R. & von Spiczak, S. Genetics and genetic diagnosis of focal childhood epilepsy. Clin Epileptol (2024). https://doi.org/10.1007/s10309-024-00677-6
Accepted:
Published:
DOI: https://doi.org/10.1007/s10309-024-00677-6