
Abstract
Background
Circulating tumour cells (CTCs) are attractive “liquid biopsy” candidates that could provide insights into the different phenotypes of tumours present within a patient. The epithelial-to-mesenchymal transition (EMT) of CTCs is considered a critical step in tumour metastasis; however, it may confound traditional epithelial feature-based CTC isolation and detection. We applied single-cell copy number alteration (CNA) analysis for the identification of genomic alterations to confirm the neoplastic nature of circulating cells with only mesenchymal phenotypes.
Methods
We isolated CTCs from blood samples collected from 46 NSCLC patients using the Parsortix system. Enriched cells were subjected to immunofluorescent staining for CTC identification using a multi-marker panel comprising both epithelial and mesenchymal markers. A subset of isolated CTCs was subjected to whole genome amplification (WGA) and low-pass whole-genome sequencing (LP-WGS) for the analysis of copy number alterations (CNAs).
Results
CTCs were detected in 16/46 (34.8%) patients, inclusive of CK+/EpCAM+ CTCs (3/46, 6.5%) and Vim+ CTCs (13/46, 28.3%). Clusters of Vim+ cells were detected in 8 samples, which constitutes 50% of the total number of NSCLC patients with CTCs. No patients had detectable hybrid CK+/EpCAM+/Vim+ cells. All of the tested CK+/EpCAM+ CTCs and 7/8 Vim+ CTCs or CTC clusters carried CNAs confirming their neoplastic nature. Notably, the Vim+ cluster with no CNAs was characterised by spindle morphology and, therefore, defined as normal mesenchymal circulating cells.
Conclusion
Our results revealed that CK-negative, vimentin-expressing cells represent a large proportion of CTCs detected in NSCLC patients, which are likely missed by standard epithelial-marker-dependent CTC categorisation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Lung cancer is the worldwide leading cause of cancer mortality, with an estimated number of deaths reaching 1.8 million in 2020 alone (Sung et al. 2021). Non-small cell lung cancer (NSCLC) accounts for 80–85% of lung cancer cases (Zappa and Mousa 2016). NSCLC patients are typically diagnosed at an advanced stage, and the estimated 5-year survival rate for NSCLC is 26.4% (Ganti et al. 2021). Immunotherapy has revolutionised the treatment paradigm of NSCLC, significantly prolonging the overall survival of advanced-stage patients (Chiang and Herbst 2020; Berghmans et al. 2020). Despite the striking clinical improvement with immunotherapies, the majority of patients eventually fail to respond to these drugs due to the evolution of primary or secondary resistance (Borghaei et al. 2015; Brahmer et al. 2012).
Unravelling aggressive tumour cell phenotypes as they evolve during treatment can provide predictive insights into the occurrence of resistance to standard of care treatments. Circulating tumour cells (CTCs) have emerged as a minimally invasive “liquid biopsy” strategy that has particular relevance for NSCLC due to the complexities of obtaining a lung biopsy (Alix-Panabières and Pantel 2016; Keller and Pantel 2019). CTCs are tumour-derived cells shed from diverse neoplastic deposits in the bloodstream (Keller and Pantel 2019; Alix‐Panabières and Pantel 2017). As such, CTCs mirror tumour heterogeneity of both primary tumours and their metastases, making them excellent candidates that reflect the phenotypes of all lesions present within a patient at any one time (Hanssen et al. 2015). Moreover, while it is difficult in clinical practice to obtain repeat biopsies from any lesion, CTCs acquired through a sample of blood could be used as a source of information on the tumour tissue over time (Lucci et al. 2016; Manjunath et al. 2019; Spencer 2020). This protocol allows multi-phenotypic subty** of CTCs utilising sequential fluorescent quenching and re-staining for further biomarkers (Adams et al. 2016; Acheampong et al. 2022). Following quenching of fluorescence from the initial round of immunostaining, samples were re-stained with mesenchymal markers (vimentin-AF647, and N-cadherin-PE). Slides were visualised and scanned using an Eclipse Ti-E inverted fluorescent microscope (Nikon, Chiyoda, Japan). Images were analysed using the NIS-Elements Analysis software, version 5.21 (Nikon).
Characterisation of enriched cells at the single-cell level
To confirm the malignant nature of the putative CTC populations by detecting genome-wide CNAs, the CellCelector (ALS, Jena, Germany) platform was employed to pick individual pCK and vimentin (Vim) only expressing cells from stained slides, previously optimised in our lab (Beasley et al. 2018). Picked cells were subjected to whole-genome amplification (WGA) using the Ampli1 WGA Kit (Silicon Biosystems, Bologna, Italy) according to the manufacturer’s specifications. Quality control of WGA-DNA was performed using Ampli1 QC Kit following the manufacturer’s instructions (Silicon Biosystems). WGA-DNA was used to construct 400 bp sequencing libraries using the Ampli1 LowPass Kit for Ion Torrent (Silicon Biosystems) following the manufacturer’s instructions. Pooled library was diluted to 50 pM and loaded into an Ion 530 Chip (Life Technologies) using the Ion Chef (400 base chemistry) (Life Technologies) and sequenced on an Ion S5 (Life Technologies) for 525 flows. CNAs were analysed using the Ion Reporter Software (Life Technologies).
Statistical analysis
Demographic data were presented as numbers, ranges, counts, percentages, means, and medians. The graph for CTCs counts was processed using GraphPad Prism version 8.0.2. The primary endpoints of the outcome analysis were overall survival (OS) and progression-free survival (PFS). Kaplan–Meier method was used to estimate median OS and PFS within groups and differences in patient survival rates were determined using log-rank tests. Univariate Cox regression hazard model for OS was performed for age, sex, NSCLC stage, ECOG performance status, histological type, tumour PD-L1 expression, treatment, and CTC counts. All survival analyses were performed in R version 4.05 using the package “survplot”. P values less than 0.05 were considered statistically significant.
Results
Demographics of patients
The patient demographics are summarised in Table 1. The median age of patients was 72 years. Majorities of the patients were male (65.2%), had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1 (69.5%), and were smokers (95.6%). Most patients had stage IV M1c NSCLC (60.8%), and adenocarcinoma was the most histological type (78.3%) among patients. Fourteen patients had tumours with KRAS mutations (30.4%). Information on tumour PD-L1 expression was available for most patients, with most tumour biopsies (65.2%) expressing PD-L1 in 1% or more of tumour cells.
Enrichment and detection of CTCs
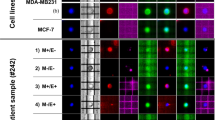
All enriched samples had two rounds of immunofluorescence staining for CTC characterisation. CTCs were initially identified as CK+/EpCAM+, DAPI+, CD45−/16−, and with or without PD-L1 expression. The fluorophores were quenched using borohydride and the samples were re-stained with vimentin and N-Cadherin. Different CK+/EpCAM+ and Vim+ expression patterns observed in CTCs from patients are shown in Fig. 1.

Representative fluorescence images of cells enriched with the Parsortix system. Cells were stained with antibodies for pan-cytokeratins and EpCAM (green), CD45 and CD16 (red), and PD-L1 (white) to identify and characterise classical NSCLC CTCs, followed by fluorescence quenching and re-staining with antibodies for N-cadherin (cyan) and vimentin (pink). Scale bar represents 10 μm
In the 46 NSCLC patients, CK+/EpCAM+ cell types were found in 3/46 (6.5%) patients’ samples (range 1–4 cells/sample) (Fig. 2). Surprisingly, none of the CTCs were positive for PD-L1 expression. However, all patients had a proportion of their WBCs expressing PD-L1 (Figure S1). Cells expressing only vimentin (CK−/EpCAM−/Vim+) with negative for CD45/16 expression were found in 13/46 (28.3%) samples (range 1–33 cells/sample). None of the patient samples had detectable hybrid CK+/EpCAM+/Vim+ CTCs. Of the 16 patients with detectable CTCs, 50% had CTC clusters all of which were vimentin-positive only. The number of cells within clusters ranged from 2 to 10 cells. All the CK+/EpCAM+ cells were present as single cells. None of the analysed patients’ samples had both CK+/EpCAM+ and Vim+ cell types. Overall, a total of 169 CTCs (range 1–33) were detected in 16/46 (34.8%) patients’ samples (Fig. 2).

CTC counts in NSCLC patients. Blood samples collected before commencing treatment were enriched for CTCs using the Parsortix system (n = 46). The number of cells on each sample is indicated on top of the bars. Bar colours indicate whether identified CTCs expressed CK/EpCAM—green, or vimentin—purple. *Samples with CTC clusters
Copy number alteration analysis of detected circulating cells from NSCLC patients
To confirm the malignancy of the Vim+ putative CTCs found as clusters or single cells, we utilised WGA and LP-WGS to detect chromosomal CNAs. Additionally, EpCAM+/CK+ CTCs found in patients 1467 and 1500 were also analysed. Overall, we obtained genomic profiles from a total of 13 CTCs or CTC clusters (Figs. 3, 4). In addition, four WBCs were also analysed as negative controls of any potential artifacts during the WGA (Figure S2). Patient 1467 had four classical CK+/EpCAM+ CTCs detected, which were picked and analysed for CNA (Fig. 3). Three of the CTCs displayed multiple CNAs and similar profiles despite some heterogeneity between cells. One of the four CTCs had low-quality sequencing and CNAs were not assessable (Fig. 3, CTC 4). The morphology of this cell suggests that it is undergoing apoptosis or has been damaged.

CNA profiles of epithelial CTCs. Immunostaining and morphology of CK+/EpCAM+ CTCs in relation to their CNA profiles obtained from low-pass whole-genome sequencing. Blue lines indicate copy gains and red lines copy number losses

CNA profiles of mesenchymal CTCs. Immunostaining and morphology of vimentin-expressing CTCs in relation to their CNA profiles obtained from low-pass whole-genome sequencing. Blue lines indicate copy gains and red lines copy number losses
We also analysed the genomic profiles of single and clusters of putative CTCs identified via vimentin expression (Fig. 4). These Vim+ cells displayed heterogeneous chromosomal CNA distribution patterns. The two single cells isolated from 1476 displayed similar CNA profiles, while the two clusters from 1472 were distinct. Only one cluster of two cells was analysed from 1383, and it was found to have CNAs. Finally, three clusters of Vim + cells were obtained from 1321; two of the clusters showed CNAs while the third displayed a ‘flat’ normal diploid CNA profile comparable to WBC controls (Figure S2). Notably, the third cluster of Vim + cells displayed a distinct spindle morphology (Fig. 4, Vim 8).
Survival analysis
We analysed the association of patient characteristics and CTC counts with clinical outcomes, OS, and PFS in participants with a minimum of 6 months of follow-up time and that were treated with systemic therapies (n = 40). A threshold of one Vim+CTC was used for survival analysis because of the low number of detected CTCs. The median follow-up for OS and PFS for the cohort were 12.3 months (95% CI 9.5–20.9) and 12.2 months (95% CI: 9.4–17.7), respectively. Log-rank survival analysis showed similar median PFS for patients with ≥ 1 Vim+ CTCs compared to patients without Vim+ CTCs. However, patients with ≥ 1 Vim+ CTCs had a median OS of 4.9 months compared to 14.5 months observed for those with no Vim+ CTCs (Fig. 5).

Kaplan–Meier curves for OS (a) and PFS (b) of patients with and without vimentin-expressing–Vim( +) CTCs. Log-rank p values, group numbers, percentages, median, and 95% CIs are indicated for each plot
Cox regression models again showed no difference in PFS (HR = 1.42, 95% CI 0.68 to − 2.94, p = 0.341), while Vim+ CTCs was associated with worse OS (HR = 1.98, 95% CI 0.89–4.45, p = 0.096), albeit not statistically significant (Fig. 5 and Table 2). There were no statistically significant associations of the patients' demographics with clinical outcomes, with regard to age, sex, ECOG status, stage of disease, tumour PD-L1 expression, and histological type (Table 2).
Discussion
Currently, the EpCAM/CK-dependent CellSearch system remains the only FDA-approved CTC enumeration platform. However, this system fails to recognise CTCs that have downregulated epithelial markers while undergoing EMT. This leads to an underestimation of the CTCs undergoing EMT with hybrid or mesenchymal phenotypes (Yin et al. 2015; Yu et al. 2015).
Our study employed the Parsortix system for CTC enrichment and an immunofluorescence protocol that combines both epithelial and mesenchymal markers for CTC identification. Our detection rate (34.8%) was considerably lower than those obtained in most of the previous studies that used the Parsortix system. Janning et al. reported a detection rate of 61% (Janning et al. 2019), and Papadaki et al., detected CTCs in 60% of total patients (Papadaki et al. 2020). However, the observed detection in this study was similar in comparison to a study by Mondelo-Macia et al., who reported a CTC detection rate of 35% in metastatic NSCLC patients using the Parsortix (Mondelo‐Macía et al. 2021). These previous studies used CK and EpCAM expression as positive markers and CD45 expression as the only negative WBC marker for CTCs. This restricted definition of CTCs does not take into account the variation in WBCs in the bloodstream with little or absent CD45 expression such as neutrophils (Gorczyca et al. 2011). Compounding the issue to a higher extent is the evidence that neutrophils stain positive for CK (Schehr et al. 2016; Streicher et al. 1998), raising concerns about the specificity of the traditional definition of CTCs. Beyond CD45, this study used CD16 for WBC identification, which has been found to significantly reduce the number of false positive CTCs (Swennenhuis et al. 2016; Spencer 2020). These factors may explain the low CTC detection rate in our study when compared to others.
One unanticipated finding was that in our cohort none of the detected NSCLC CTCs had PD-L1 expression, despite 65.2% of the cohort having PD-L1-positive tumours (> 1%), with 36.5% with more than 50% tumour specific staining. Published studies have consistently reported wide ranges of the rate of PD-L1 expressing CTCs, from 8 to 100%, in NSCLC (Acheampong et al. 2020; Kong et al. 2021; Ouyang et al. 2021). Detection of PD-L1 expression is influenced by a range of different factors including different antibodies, cut-off values, and CTC isolation platforms (Ouyang et al. 2021; Acheampong et al. 2020). Nevertheless, we detected PD-L1 expression on a subset of WBCs present in the enriched samples which is consistent with reports from previous studies (Ilié et al. 2018; Kotsakis et al. 2019).
EpCAM and different members of the cytokeratin family are frequently utilised for CTC identification before subsequent characterisation due to the epithelial nature of NSCLC (Hamilton and Rath 2016). Interestingly, the majority of the detected circulating cells in this study expressed vimentin with total loss of EpCAM and cytokeratin. More precisely, many of these Vim+ cells had cytomorphological characteristics such as shape and size consistent with CTCs, in addition to being negative for WBC markers (Boffa et al. 2017). Moreover, all putative NSCLC CTCs in this study were negative for N-cadherin. Since these identified circulating cells do not meet the field consensus criteria for CTCs, due to their lack of CK we assessed the neoplastic origin of these Vim+ cells together with CK+/EpCAM+ CTCs by LP-WGS analysis.
Classical CTCs presented with significant genomic alterations with some degree of heterogeneity. Among the Vim+ cells, those presenting CNAs could be considered bona fide CTCs. Our findings are in line with a previous study by Xu et al. who identified Vim+ circulating cells in metastatic prostate cancer patients using the Parsortix and confirmed their malignancy by genomic alterations (Xu et al. 2017). Another study by Reduzzi et al. demonstrated that enriched circulating cells lacking both epithelial and leukocyte marker expression presented altered CNA profiles and thus were defined as CTCs (Reduzzi et al. 2020). Because the authors only identified double negative cells for the CNA analysis, the actual phenotype of these cells was unknown. By contrast, our putative CTCs were phenotypically identified to be Vim+/CK− cells (Reduzzi et al. 2020). These reports together indicate the relevance of the existence of CTCs subpopulations that express low or no EpCAM or CK and can escape detection by epithelial isolation methods and markers.
It is also worth noting that some Vim+ cells did not carry CNA and were, therefore, classified as non-tumourigenic mesenchymal-derived circulating cells. Vimentin is also expressed in circulating endothelial cells, which have been shown to also increase in cancer patients (Chen et al. 2021; Lin et al. 2017). The presence of hematopoietic cells of mesenchymal origin and reactive stromal cells compromise the specificity for recognition of CTCs via mesenchymal markers (Stoecklein et al. 2016; Schehr et al. 2016; Cima et al. 2016; Plaks et al. 2013). Preferably, additional exclusion markers should be included in future panels. Cancer patients have been shown to have an increased number of circulating endothelial cells (CECs) that are probably shed from tumour angiogenesis-associated processes or damaged tumour vessel walls (Ilie et al. 2014). Therefore, immunoassays for CTCs should include markers such as CD31, CD34, and CD144 to exclude cells of endothelial origin (Magbanua et al. 2015; Bidard et al. 2010).
The prognostic value of vimentin expression in various cancers has been well documented in the literature (Santamaria et al. 2017; Dongre and Weinberg 2019). High levels of vimentin expression are associated with poor survival in patients with NSCLC (Ye et al. 2016). Although our survival analyses in patients with Vim+ CTCs did not achieve statistically significant levels, they showed a trend towards unfavourable OS. A previous study by Zhang et al. indicated mesenchymal CTCs identified NSCLC patients with distant metastasis (Zhang et al. 2019).
Our study employed a robust CTC isolation, identification, and downstream single-cell genomic analysis. Nonetheless, some limitations need to be mentioned. The sample size of the study was small. We were not able to compare the CNA profiles of NSCLC CTCs to that of the matching tumours as samples were not available for profiling. Moreover, some Vim+ cells collected could not be used for CNA analysis due to unsuccessful amplification. This may have been caused by the method used to fix and immunostained the cells, which may have compromised the quality of the nucleus, or our inability to effectively recover the nuclei using the micromanipulator (CellCelector).
Conclusion
Overall, these results reveal that pCK negative, EpCAM negative, and vimentin expressing cells represent a large proportion of CTCs detected in NSCLC patients. These cells would be commonly missed by standard CTC categorisation. The presence of CNAs confirmed the neoplastic nature of a proportion of vimentin-only expressing cells. The lack of CNA in some of the CK− Vim+ cells underscores the need for better markers to identify and quantify CTCs. Future longitudinal studies with larger cohorts are needed to validate the clinical validity of epitope-independent microfluidic technologies for CTC isolation and the consideration of the addition of Vim+ CTCs to the classical CTC definition in NSCLC and evaluation of their prognostic value.
Data availability
Raw data are available upon request.
References
Acheampong E, Abed A, Morici M, Bowyer S, Amanuel B, Lin W, Millward M, Gray ES (2020) Tumour PD-L1 expression in small-cell lung cancer: a systematic review and meta-analysis. Cells 9:2393
Acheampong E, Abed A, Morici M, Spencer I, Beasley AB, Bowyer S, Asante D-B, Lomma C, Lin W, Millward M (2022) Evaluation of PD-L1 expression on circulating tumour cells in small-cell lung cancer. Transl Lung Cancer Res 11:440
Adams DL, Katherine Alpaugh R, Tsai S, Tang C-M, Stefansson S (2016) Multi-Phenotypic subty** of circulating tumor cells using sequential fluorescent quenching and restaining. Sci Rep 6:33488
Alix-Panabières C, Pantel K (2016) Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 6:479–491
Alix-Panabières C, Pantel K (2017) Characterization of single circulating tumor cells. FEBS Lett 591:2241–2250
Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR (2014) Epithelial–mesenchymal transition in lung development and disease: does it exist and is it important? Thorax 69:760–765
Beasley A, Isaacs T, Khattak MA, Freeman JB, Allcock R, Chen FK, Pereira MR, Yau K, Bentel J, Vermeulen T (2018) Clinical application of circulating tumor cells and circulating tumor DNA in uveal melanoma. JCO Precis Oncol 2:1–12
Berghmans T, Durieux V, Hendriks LEL, Dingemans A-M (2020) Immunotherapy: from advanced NSCLC to early stages, an evolving concept. Front Med 7:90
Bidard F-C, Mathiot C, Armelle Degeorges M-C, Etienne-Grimaldi RD, Xavier Pivot C, Veyret LB, de Cremoux P, Milano G (2010) Clinical value of circulating endothelial cells and circulating tumor cells in metastatic breast cancer patients treated first line with bevacizumab and chemotherapy. Ann Oncol 21:1765–1771
Boffa DJ, Graf RP, Salazar MC, Hoag J, David Lu, Krupa R, Louw J, Dugan L, Wang Y, Landers M (2017) Cellular expression of PD-L1 in the peripheral blood of lung cancer patients is associated with worse survival. Cancer Epidemiol Prev Biomark 26:1139–1145
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E (2015) Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med 373:1627–1639
Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Kunle J (2012) Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N England J Med Odunsi 366:2455–2465
Chen Z, Fang Z, Ma Ji (2021) Regulatory mechanisms and clinical significance of vimentin in breast cancer. Biomed Pharmacother 133:111068
Chiang AC, Herbst RS (2020) Frontline immunotherapy for NSCLC—the tale of the tail. Nat Rev Clin Oncol 17:73–74
Chudziak J, Burt DJ, Mohan S, Rothwell DG, Mesquita B, Antonello J, Dalby S, Ayub M, Priest L, Carter L (2016) Clinical evaluation of a novel microfluidic device for epitope-independent enrichment of circulating tumour cells in patients with small cell lung cancer. Analyst 141:669–678
Ciccioli M, Bravo-Santano N, Davis A, Lewis J, Malcolm R, Pailhes-Jimenez A-S (2021) Abstract 588: Mesenchymal markers: The new avenue for circulating tumor cells detection. Cancer Res 81(13_Supplement):588. https://doi.org/10.1158/1538-7445.Am2021-588
Cima I, Say LK, Debarka S, Iain BT, Wai MP, Daniel L, Min H, Ciprian I, Irina A, Wei LG (2016) Tumor-derived circulating endothelial cell clusters in colorectal cancer. Sci Transl Med 8(345):ra89–ra4589
Cote RJ, Ram HD (2016) Circulating tumor cells, 1st edn. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-3363-1
Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20:69–84
Ganti AK, Klein AB, Cotarla I, Seal B, Chou E (2021) Update of incidence, prevalence, survival, and initial treatment in patients with non–small cell lung cancer in the US. JAMA Oncol 7:1824–1832
Gorczyca W, Sun Z-Y, Cronin W, Li X, Mau S, Tugulea S (2011) Immunophenotypic pattern of myeloid populations by flow cytometry analysis. Methods Cell Biol 103:221–266
Hamilton G, Rath B (2016) Detection of circulating tumor cells in non-small cell lung cancer. J Thorac Dis 8:1024
Hanssen A, Loges S, Pantel K, Wikman H (2015) Detection of circulating tumor cells in non-small cell lung cancer. Front Oncol 5:207
Hyuna S, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin 71:209–249
Ilie M, Long E, Hofman V, Selva E, Bonnetaud C, Boyer J, Vénissac N, Sanfiorenzo C, Ferrua B, Marquette CH (2014) Clinical value of circulating endothelial cells and of soluble CD146 levels in patients undergoing surgery for non-small cell lung cancer. Br J Cancer 110:1236–1243
Ilié M, Szafer-Glusman E, Hofman V, Chamorey E, Lalvée S, Selva E, Leroy S, Marquette C-H, Kowanetz M, Hedge P (2018) Detection of PD-L1 in circulating tumor cells and white blood cells from patients with advanced non-small-cell lung cancer. Ann Oncol 29:193–199
Janning M, Kobus F, Babayan A, Wikman H, Velthaus J-L, Bergmann S, Schatz S, Falk M, Berger L-A, Böttcher L-M (2019) Determination of PD-L1 expression in circulating tumor cells of NSCLC patients and correlation with response to PD-1/PD-L1 inhibitors. Cancers 11:835
Keller L, Pantel K (2019) Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat Rev Cancer 19:553–567
Kong D, Zhang W, Yang Z, Li G, Cheng S, Zhang K, Feng L (2021) Correlation between PD-L1 expression ON CTCs and prognosis of patients with cancer: a systematic review and meta-analysis. Oncoimmunology 10:1938476
Kotsakis A, Kallergi G, Aggouraki D, Lyristi Z, Koinis F, Lagoudaki E, Koutsopoulos A, Georgoulias V, Vetsika E-K (2019) CD8+ PD-1+ T-cells and PD-L1+ circulating tumor cells in chemotherapy-naïve non-small cell lung cancer: towards their clinical relevance? Therapeutic Adv Med Oncol 11:1758835919853193
Krebs MG, Hou J-M, Sloane R, Lancashire L, Priest L, Nonaka D, Ward TH, Backen A, Clack G, Hughes A (2012) Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and-independent approaches. J Thorac Oncol 7:306–315
Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, Farace F (2011) Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer 105:1338–1341
Li J, Liao Yi, Ran Y, Wang G, Wei Wu, Qiu Y, Liu J, Wen N, **g T, Wang H (2020) Evaluation of sensitivity and specificity of CanPatrol™ technology for detection of circulating tumor cells in patients with non-small cell lung cancer. BMC Pulm Med 20:1–9
Lin PP, Gires O, Wang DD, Li L, Wang H (2017) Comprehensive in situ co-detection of aneuploid circulating endothelial and tumor cells. Sci Rep 7:1–10
Lucci A, Hall CS, Lodhi AK, Bhattacharyya A, Anderson AE, **ao L, Bedrosian I, Kuerer HM, Krishnamurthy S (2012) Circulating tumour cells in non-metastatic breast cancer: a prospective study. Lancet Oncol 13:688–695
Magbanua MJM, Michael P, ** SL, Marc J, Victoria W, Matthew G, Karen M, Julia P, Harwinder S, Arejas U (2015) A novel strategy for detection and enumeration of circulating rare cell populations in metastatic cancer patients using automated microfluidic filtration and multiplex immunoassay. PLoS ONE 10:e0141166
Maly V, Ondrej M, Katarina K, Vladimir B (2019) Circulating tumor cells in diagnosis and treatment of lung cancer. In Vivo 33:1027–1037
Manjunath Y, Upparahalli SV, Avella DM, Deroche CB, Kimchi ET, Staveley-Ocarroll KF, Charles JS, Guangfu L, Jussuf TK (2019) PD-L1 expression with epithelial mesenchymal transition of circulating tumor cells is associated with poor survival in curatively resected non-small cell lung cancer. Cancers 11:806
Mondelo-Macía P, García-González J, León-Mateos L, Anido U, Aguín S, Abdulkader I, Sánchez-Ares M, Abalo A, Rodríguez-Casanova A, Díaz-Lagares Á (2021) Clinical potential of circulating free DNA and circulating tumour cells in patients with metastatic non-small cell lung cancer treated with pembrolizumab. Mol Oncol 15:2923–2940
Ouyang Y, Liu W, Zhang N, Yang X, Li J, Long S (2021) Prognostic significance of programmed cell death-ligand 1 expression on circulating tumor cells in various cancers: a systematic review and meta-analysis. Cancer Med 10:7021–7039
Papadaki MA, Sotiriou AI, Vasilopoulou C, Filika M, Aggouraki D, Tsoulfas PG, Apostolopoulou CA, Rounis K, Mavroudis D, Agelaki S (2020) Optimization of the enrichment of circulating tumor cells for downstream phenotypic analysis in patients with non-small cell lung cancer treated with anti-pd-1 immunotherapy. Cancers 12:1556
Plaks V, Koopman CD, Werb Z (2013) Circulating tumor cells. Science 341:1186–1188
Reduzzi C, Vismara M, Silvestri M, Celio L, Niger M, Peverelli G, De Braud F, Daidone MG, Cappelletti V (2020) A novel circulating tumor cell subpopulation for treatment monitoring and molecular characterization in biliary tract cancer. Int J Cancer 146:3495–3503
Santamaria PG, Moreno-Bueno G, Portillo F, Cano A (2017) EMT: present and future in clinical oncology. Mol Oncol 11:718–738
Schehr JL, Schultz ZD, Warrick JW, Guckenberger DJ, Pezzi HM, Sperger JM, Heninger E, Saeed A, Leal T, Mattox K (2016) High specificity in circulating tumor cell identification is required for accurate evaluation of programmed death-ligand 1. PLoS ONE 11:e0159397
Spencer I (2020) Characterising PD-L1 expression in circulating melanoma and non-small cell lung cancer cells. https://ro.ecu.edu.au/theses/2318
Stoecklein NH, Fischer JC, Niederacher D, Terstappen LWMM (2016) Challenges for CTC-based liquid biopsies: low CTC frequency and diagnostic leukapheresis as a potential solution. Expert Rev Mol Diagn 16:147–164
Streicher J, Fabian B, Herkner K, Pointner H, Bayer PM (1998) Anticytokeratins are a potential source of false-positive indirect immunofluorescence assays for C-ANCA. J Clin Lab Anal 12:54–59
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/caac.21660
Swennenhuis JF, van Dalum G, Zeune LL, Terstappen LWMM (2016) Improving the cell search® system. Expert Rev Mol Diagn 16:1291–1305
Tania M, Khan MA, Junjiang Fu (2014) Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumor Biology 35:7335–7342
Xu L, Mao X, Imrali A, Syed F, Mutsvangwa K, Berney D, Cathcart P, Hines J, Shamash J, Yong-Jie Lu (2015) Optimization and evaluation of a novel size based circulating tumor cell isolation system. PLoS ONE 10:e0138032
Xu L, Mao X, Guo T, Chan PY, Shaw G, Hines J, Stankiewicz E, Yuqin Wang R, Oliver TD, Ahmad AS (2017) The novel association of circulating tumor cells and circulating megakaryocytes with prostate cancer prognosis. Clin Cancer Res 23:5112–5122
Ye Z, Zhang X, Luo Y, Li S, Huang L, Li Z, Li P, Chen G (2016) Prognostic values of vimentin expression and its clinicopathological significance in non-small cell lung cancer: a meta-analysis of observational studies with 4118 cases. PLoS ONE 11:e0163162
Yin J, Wang Yi, Yin H, Chen W, ** G, Ma H, Dai J, Chen J, Jiang Y, Wang H (2015) Circulating tumor cells enriched by the depletion of leukocytes with bi-antibodies in non-small cell lung cancer: potential clinical application. PLoS ONE 10:e0137076
Yu Na, Zhou J, Cui F, Tang X (2015) Circulating tumor cells in lung cancer: detection methods and clinical applications. Lung 193:157–171
Zappa C, Mousa SA (2016) Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res 5:288
Zhang X, Wei L, Li J, Zheng J, Zhang S, Zhou J (2019) Epithelial-mesenchymal transition phenotype of circulating tumor cells is associated with distant metastasis in patients with NSCLC. Mol Med Rep 19:601–608
Acknowledgements
We thank healthy volunteers and patients for the provision of samples for this study. We also acknowledge the phlebotomy and nursing staff of participating hospitals in acquiring blood samples. We express our gratitude to our team members, Anna Reid, Isaac Spencer, and Paula Van Miert that helped with sample collection and transportation.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. E.A. and D.A were supported by Australian Government Research Training Program (RTP) Scholarships. E.S.G. was supported by a fellowship from the Cancer Council of Western Australia. The study was partially supported by the Cancer Research Trust ‘Enabling advanced single-cell cancer genomics in WA’ grant and the Centre for Precision Health High Degree Research student grant scheme 2021.
Author information
Authors and Affiliations
Contributions
EA and ES designed the study; EA, MMo, and ABB carried out experimental work; MMo and DA provided protocol details and critical advice; EA and ABB analysed data and statistical analysis; AA and SB enrolled patients in the study, collected and curated clinical data; AA, WL, MMi, and ES supervised the study. EA drafted the manuscript. All authors read and approved the final version of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that no competing interest exists.
Ethical approval and consent to participate
Written informed consent was obtained from all patients and procedures were approved by Human Research Ethics Committees at Edith Cowan University (No.18957) and Sir Charles Gairdner Hospital (No. 2013-246 and RGS0000003289) in compliance with Helsinki Declaration. Experiments were conducted per institutional and national guidelines and regulations.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Acheampong, E., Morici, M., Abed, A. et al. Powering single-cell genomics to unravel circulating tumour cell subpopulations in non-small cell lung cancer patients. J Cancer Res Clin Oncol 149, 1941–1950 (2023). https://doi.org/10.1007/s00432-022-04202-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04202-y