
Abstract
Atrial fibrillation (AF) occurs from disordered atrial action potential conduction and is associated with reduced gap junction electrical conductance (Gj). The Ca2+ and calmodulin-dependent phosphatase, calcineurin, reduces Gj in ventricular myocardium via a protein phosphatase-1 (PP1)-dependent pathway culminating in phosphorylation of serine368 on connexin43 (pSer368-Cx43). However, characterisation of corresponding pathways in left atrial myocardium, which have a more complex connexin subtype profile, is undefined and was the aim of this study. Gj was measured in guinea-pig left atrium from the frequency-dependent variation of intracellular impedance; intracellular [Ca2+], ([Ca2+]i) in low-Na solution was measured by Fura-2 fluorescence. Phosphorylation of guinea-pig Ser368-Cx43 residues was measured by Western blot; Cx40 was immunoprecipitated and probed for serine/threonine residue phosphorylation. Low-Na solution reversibly reduced Gj, in turn attenuated or prevented by calcineurin inhibitors cyclosporin-A or CAIP, respectively. Moreover, Ser368-Cx43 phosphorylation in low-Na solution was also prevented by CAIP. Changes were partially prevented by fostreicin (FST), a protein phosphatase-2A (PP2A) inhibitor; but not by tautomycin, a PP1 inhibitor. Serine/threonine residues on Cx40 were also phosphorylated in low-Na solution; prevented by CAIP and attenuated by FST. Reduced Gj with raised [Ca2+]i is paralleled by a changed Cx43/Cx40 phosphorylation status; changes mediated by calcineurin and PP2A-dependent pathways, but not PP1. The pharmacological profile underlying changes to guinea-pig atrial gap junction electrical conductance with raised intracellular [Ca2+]i is fundamentally different from that in ventricular myocardium. This provides a targeted drug model whereby atrial and ventricular myocardium can be selectively targeted to correct conduction defects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) remains a major cause of morbidity and mortality, including risk of thromboembolism and cerebrovascular stroke [1]. A rise of the sarcoplasmic [Ca2+] ([Ca2+]i) is a key contributor to initiation of AF [2] and consequential generation of re-entrant rhythms that require a region of slow action potential (AP) propagation. The pathway whereby raised [Ca2+]i reduces AP propagation is less clear, although a key factor is the electrical conductance (Gj) offered by gap junctions between contiguous myocytes [3]. Gap junctions (GJ) in atrial myocardium are composed of connexin (Cx) phosphoproteins, with two main isoforms, Cx40 and Cx43, that form homotypic or heterotypic structures, each with a characteristic unit value of Gj [4]. Overall Gj and AP propagation velocity will depend on the relative expression of Cx isoforms [5, 6].
Moreover, the phosphorylation state of several serine (Ser) and threonine (Thr) residues on Cx isoforms determines unit Gj and the role of Ser/Thr protein kinases (PK) and phosphatases (PP) are crucial [7, 8]. Ca2+/calmodulin-dependent PKII (CaMKII) phosphorylation targets on Cx43 have been extensively mapped [9]. However, only serine-368 (Ser368) Cx43 phosphorylation, targeted by PKC and which decreases Gj, has been characterised [10]. Access to the Ser368-Cx43 residue is by dephosphorylation of a gatekeeper residue Ser365-Cx43 [11].
Under physiological conditions Ser365-Cx43 is in a phosphorylated state (pSer365-Cx43) and Ser368-Cx43 is poorly phosphorylated to maintain a relatively high Gj. We showed in guinea-pig ventricular myocardium [12] that raised [Ca2+]i is accompanied by slowed action potential conduction and reduced Gj, accompanied by relative dephosphorylation of Ser365-Cx43 as well as phosphorylation of Ser368-Cx43. The changed phosphorylation state of Ser365-Cx43 is dependent on the activity of the Ca2+/CaM-dependent Ser/Thr PP, calcineurin (Cn), with subsequent PKC-dependent Ser368-Cx43 phosphorylation [12]. The action of calcineurin to initiate this process is indirect, via its activation of the Ca2+-independent phosphatase PP1. However, there was no functional role for the Ca2+-independent phosphatase PP2A.
With atrial myocardium less is known about the Ca2+-dependent regulation of Gj through changes to Cx phosphorylation and the role of Cn. Moreover, there is an additional complication of two predominant atrial Cx isoforms, Cx43 and Cx40. Less is known about the relationship between phosphorylation sites and electrical conductance of Cx40 gap junctions although a potential role is suggested from changes to Cx40 homotypic Gj by raised intracellular cAMP or β-adrenoreceptor activation [13, 14].
This study aimed to answer the following questions in left atrial myocardium: does raised [Ca2+]i reduce Gj; is there a role for calcineurin in any change to Gj; what are the contributions of PP1 and PP2A to any calcineurin-dependent pathway; and are there phosphorylation changes to Cx43 and Cx40 that accompany changes to Gj. Phosphorylation status of Cx43 was assayed as the relative changes at Ser368; however, as sites affecting Gj on Cx40 are less well understood changes to total serine or threonine phosphorylation were measured.
Methods
Tissue sources and ethics
Dunkin-Hartley male guinea-pigs (300–400 g) were heparinised (100 units, i.p.), euthanised by cervical dislocation—confirmed by absent corneal and spinal reflexes—the heart excised and the left atrium dissected in Tyrode’s solution. Experimental protocols adhered to ARRIVE guidelines [15]; begun at Surrey University and finalised at the University of Bristol. Animals were procured by the local animal services units, housed in straw-floored plastic cages at 22 °C with a 12-h light–dark cycle and with water and food available ad libitum.
Measurement of gap junction conductance
Left atrial strips (< 1.0 mm diam, ≈5 mm length) were dissected immediately after atrial excision and mounted in a three-chamber Perspex bath at 37 °C. Chambers were separated by thin rubber membranes and the strip pulled gently through holes in the membranes so as to span the three chambers. Outer chambers were superfused with Tyrode’s and the central chamber filled with mineral oil. Alternating current (a.c.; 0.1–100 kHz; 10 mV peak-to-peak) was passed between the two outer chambers via Pt-black electrodes. This experimental system constrained a.c. to pass through the intracellular pathway of the preparation, with a small parallel resistance shunt, rec, from Tyrode’s solution trapped in the extracellular space. This system formed one arm of an a.c. (Wien) Bridge arrangement (Wayne-Kerr 6425, Bognor Regis, UK) to allow total impedance (z) to be recorded as a function of a.c. frequency. The method and its validation have been described in detail [5, 16, 17]. Pt-black electrode impedance, zelec, in series with the preparation in the oil gap, was measured separately in a large volume of Tyrode’s solution; extracellular fluid resistance, rec, in parallel to total intracellular impedance (zi), was also measured separately as the resistance between two needle Pt-black electrodes placed in the muscle within the central oil gap. Subtraction of zelec and rec from total impedance, z, yielded a value of left atrial zi over this range of frequencies. Values of zi were transformed to yield a frequency-independent sarcoplasmic conductance (gc), and an in-series frequency-dependent gap junction conductance (gj). Two sets of recordings of z were made for each intervention, five minutes apart; values that were within 5% were averaged and retained for analysis. After control recordings in Tyrode’s solution the mineral oil was removed, the preparation exposed to a gassed intervention solution for 20 min before the oil was re-applied and new measurements made. Finally, preparation length and diameter in the oil gap were measured to yield ‘specific’ gap junction conductance, Gj (mS.cm−2) values, i.e. independent of preparation dimensions.
Histology
Atrial strips as used for impedance measurements were snap-frozen in liquid N2, immediately after the heart was removed and dissected (t = 0) or after 45 min (t = 45) in an oil-gap chamber and stored at –80 °C. Frozen sections (12 μm) were fixed with ice-cold 4% paraformaldehyde (15 min at room temperature) and stained with haematoxylin and eosin. Sections were mounted and viewed at × 40 magnification.
Measurement of [Ca2+]i
Large myocyte clusters were prepared by collagenase dispersion of the left atrium and loaded with 5 µM Fura2-AM. [Ca2+]i was recorded as R340/380, the fluorescence emission (410–500 nm) ratio with illumination successively (32 Hz) at 340 or 380 nm. Output data were transformed through a third-order Butterworth band-pass filter centred on 32 Hz.
Western blotting
Guinea-pig hearts were perfused through the coronary vasculature for 10 min with Tyrode’s or low-Na Tyrode’s solutions in the absence or presence of phosphatase inhibitors. The left atrium was then rapidly snap-frozen in liquid N2. A whole-tissue homogenate was prepared using radio-immunoprecipitation assay (RIPA) lysis buffer, supplemented with MS SAFE protease inhibitors (Sigma, UK). Protein (30 μg) from each sample was prepared in NuPAGE® LDS sample buffer 4X with NuPAGE® sample-reducing agent 10X (Invitrogen, UK). Protein samples were resolved by 12% polyacrylamide SDS-PAGE and transferred to PVDF membranes (Invitrogen, UK), blocked with Odyssey blocking buffer (Li-COR Biosciences, Ltd, UK; 2 h; RT) and incubated overnight at 4 °C with primary antibody in 1% BSA-Tris buffered saline-Tween (TBS-T). Membranes were washed with TBS-T and incubated with secondary antibody (1:10000 dilution; 1 h, RT). Protein bands were imaged using an Odyssey infra-red (IR) imaging system (UK); red and green channels. Densitometric analyses used colour filters on Image-J software. Cx43 generally appeared as several separate bands (see Fig. 2C) and the whole set of bands were included in the densiometric analysis for total Cx43 (T-Cx43). Band densities of phosphorylated s368-Cx43 protein were normalised to total Cx43 (T-Cx43) densities and T-Cx43 normalised to GAPDH. Immunoprecipitated Cx40 (IP-Cx40. below) was probed for serine/threonine phosphorylation and band densities were also normalised to their corresponding T-Cx40 values.
Immunoprecipitation
This was undertaken to analyse the phosphorylation profile of Cx40 under different interventions, as antibodies to specific phosphorylated residues were unavailable, as they were available for Cx43 (e.g. phosphorylated serine368-Cx43). Protein G Sepharose 4 Fast Flow beads (25–50 µl bead slurry; binding capacity ≥ 20 µg IgG/µl slurry; GE Healthcare Life Sciences, UK) were conjugated with Cx antibodies (10 µl, containing 2 µg IgG) then washed with ice-cold phosphate-buffered saline (PBS) to remove unbound antibodies, as per supplier’s protocol. A protein aliquot (100 µg) from a frozen tissue sample was added to the conjugated beads and incubated overnight at 4 °C. Unbound protein was extracted and washed with PBS to remove residual protein. Beads were then incubated (95°C; 5 min) with NuPAGE® LDS sample buffer 4X and NuPAGE® sample-reducing agent 10x. After centrifugation, unbound and bound samples were processed by Western blot for total-Cx40, or phosphoserine/ phosphothreonine protein; the latter were normalised to total-Cx40 values.
Solutions and antibodies
Tyrode’s solution contained (mM): NaCl 118; KCl 4.0; NaHCO3 24; MgCl2 1.0; CaCl2 1.8; NaH2PO4 0.4; glucose 6.1; Na pyruvate 5.0; gassed with 95%O2/5%CO2, pH 7.40 ± 0.05. Low-Na Tyrode’s (29.4 mM Na), to raise the intracellular [Ca2+] ([Ca2+]i) was similar except that NaCl was replaced by TrisCl (pH to 7.4 with 1 M NaOH). The following inhibitors were used: two Cn inhibitors: i) cyclosporin-A (CysA, 5 µM; 10 mM DMSO stock), ii) Cn autoinhibitory peptide (CAIP, 50 µM; 100 mM aqueous stock) a highly-selective, cell-permeable peptide. Other phosphatase inhibitors were: a PPI inhibitor, tautomycin (TTM, 5 nM; 65.2 µM PBS stock) and a PP2A inhibitor, fostriecin (FST, 100 nM; 20 mM DMSO stock). CaMKII inhibitors were KN-93 (5 µM; 50 mM DMSO stock) and AIP (10 µM; 500 µM aqueous stock). A PKC inhibitor was chelerythrine (2 µM: 10 mM aqueous stock). The gap junction blocker, heptanol [16, 18] was used as a 1 mM solution in Tyrode’s solution. Reagents were from Sigma-Aldrich, UK, except for CysA (Calbiochem, UK), as well as AIP and FST (Tocris, UK).
Primary antibodies were: anti-total Cx43 (1:1000, rabbit polyclonal, Santa Cruz Biotechnology UK, sc-9059); anti-pS368-Cx43 (1:1000, goat polyclonal, Santa Cruz, sc-25165); anti-total Cx40 (1:1000, goat polyclonal Santa Cruz, sc-20466); anti-GAPDH (1:1000, mouse monoclonal, Santa Cruz, sc-365062); anti-phosphoserine (1:1000, mouse monoclonal, Abcam, ab7851); anti-phosphothreonine (1:1000, rabbit polyclonal, Abcam, ab9337). Secondary antibodies (1:10,000 dilution, Li-COR Biosciences Ltd, UK) were: IRDye-680 donkey anti-mouse, IRDye 680 donkey anti-goat, IRDye-800 donkey anti-rabbit and IRDye-800 donkey anti-goat antibodies. Primary antibodies were tested against left ventricle and brain lysates to find optimal dilutions.
Statistical analyses and curve-fits
Electrophysiological, Ca2+ and Western blot data are quoted as means ± SD, except where specifically designated; when several values under a particular condition were collected from a sample, these were averaged before statistical analyses, n = number of separate hearts. Comparisons between continuous data sets were tested by two-way ANOVA, with post hoc parametric tests. The null hypothesis was rejected at p < 0.05. Statistical tests used Excel and www.vassarstats.net. Impedance locus (R vs -X) plots were fitted to the equation of a circle with a radius, r, centre displaced, a, along the abscissa, (R-a)2 + (-X)2 = r2; where R is resistance and -X is reactance in the complex plane. Non-linear curve-fits used a Levenberg–Marquardt algorithm (KaleidaGraph, Synergy Software, Reading, PA, USA).
Results
Tissue viability and intracellular [Ca2+], [Ca2+]i, measurements
Tissue structural integrity over the experimental time-course (up to 40 min) was verified by fixing specimens either immediately after the heart was retrieved (t = 0), or after 40 min (t = 45) in an impedance chamber and staining with H&E (Supplemental Fig. 1A, n = 4 in total). Previous experiments also showed that tissue ATP was maintained over this time frame [18]. With large clusters of myocytes [Ca2+]i was raised (increase of Fura-2 R340/380 ratio) by low-Na superfusion and sustained for a period similar to the time-course of an experiment to measure Gj (Supplemental Fig. 1B). The resting R340/380 ratio in control was unaffected by cyclosporin-A (5 µM, CysA; n = 6), or CAIP (50 µM, n = 4). In addition, the increase in low-Na solution was similar in all three conditions (∆R340/380 ratio: 0.092 ± 0.030; 0.103 ± 0.015; 0.106 ± 0.008, respectively; p > 0.05, 2-way ANOVA).
Gap junction conductance, Gj: effect of low-Na solution; controls
Figure 1A shows an example of resistance (R) and reactance (-X) complex plane plots in Tyrode’s solution (closed circles), on exposure to low-Na solution (closed squares) and then on return to Tyrode’s solution (open circles). The intercepts of the circle fits with the abscissa are functions of sarcoplasmic resistivity, Rc (R1) and total intracellular resistivity, Ri (R2): gap junction resistivity, with Rj as Ri-Rc. Low-Na solution reversibly increased Ri, but not Rc, and hence reversibly increased Rj. Henceforth, Rj and Rc values (units Ω.cm) are expressed as inverse specific resistivity or conductance values (Gj, Gc, units mS.cm−1).

Gap junction electrical properties in control and low-Na solutions. A Plots of calculated resistance (R) and reactance (-X) in the complex plane from an individual experiment in control solution (closed circles); low-Na solution (closed squares); washout to control solution (open circles). Fits to the data are made with a circle function with a centre of radius shifted along the R-axis (see Methods). The intercepts with the abscissa, R1 and R2, are used to estimate sarcoplasmic resistivity, Rc, and intracellular resistivity, Rj, from which gap junction resistivity, Rj, is derived. B Values of Gj (= 1/Rj) in control solution, in low-Na solution and after washout to control solution. The connecting bars are data from individual experiments. Horizontal bars in each data set show median values; note the logarithmic scale of the ordinate
Low-Na solution consistently and reversibly reduced Gj to 4.5 ± 1.9 mS.cm−1, compared to pre- and post-intervention values of 8.4 ± 3.7, 8.5 ± 3.6 mS.cm−1 respectively (p < 0.001; n = 40, Fig. 1B): these two values in control solution were not significantly different (p > 0.05), with the post-intervention value 101.4 ± 8.1% of the pre-intervention value. The percentage reduction of Gj in low-Na solution was similar between preparations (54.3 ± 11.4% pre-control, p < 0.0001) and independent of the initial control value (r = 0.025).
Sarcoplasmic electrical conductance (Gc) was similar in Tyrode’s and low-Na solutions: 7.3 ± 2.2 and 6.7 ± 2.5 mS.cm−1 (n = 40; p < 0.05). Values were unaffected by any interventions and are not further reported. In Tyrode’s solution the electrical conductances offered by the sarcoplasm and gap junctions are thus similar – see Discussion for biophysical implications.
Heptanol (1 mM in Tyrode’s) reduces gap junction conductance [16, 18] and was tested in separate preparations. Sarcoplasmic conductance was unaffected by heptanol (Gc 7.8 ± 1.60 vs 7.9 ± 1.57 mS.cm−1, p > 0.05, n = 5; control vs heptanol) whereas gap junction conductance was significantly decreased (Gj 9.2 ± 1.5 vs 4.5 ± 0.4 mS.cm−1, p < 0.01, n = 5).
Gap junction conductance and ser368-Cx43 phosphorylation – role of calcineurin inhibitors
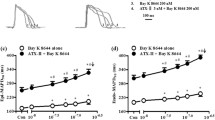
The reduction of Gj by low-Na solution was significantly attenuated in the presence of CysA (76.9 ± 9.8% of control vs 55.5 ± 9.5%; n = 6; p < 0.01); Fig. 2A, although not fully recovered compared to control levels (p < 0.001). With the more selective calcineurin inhibitor, CAIP, Gj was unaffected compared with control levels in low-Na solution (96.2 ± 5.4% of control vs 60.9 ± 7.1%; n = 6; p = 0.001); Fig. 2B.

Calcineurin inhibitors (CysA and CAIP) on gap junction conductance, Gj, and Cx43 phosphorylation. A Gj values in control or low-Na solution, each in the absence or presence of cyclosporin-A (CysA, n = 6). Gj is expressed as a percentage of values in control solution. B As with part A but with the more specific calcineurin inhibitor, CAIP (n = 6). C: Representative Western blots for total Cx43 (T-Cx43, upper blot) and pSer368-Cx43 expression (lower plot) and also, in each case, GAPDH – see Methods for details. Lane 1 is molecular weight markers; Lanes 2 and 3 are from control tissue derived protein, lanes 4–9 lysates from low-Na treated tissues (lanes 6–7 and 8–9, are also plus CysA and CAIP, respectively). D: signal quantitation for pSer368-Cx43, normalised to T-Cx43. Data are mean ± SD, all n = 6. ***p < 0.001; low-Na (± calcineurin inhibitor) vs control: §§p < 0.01, low-Na vs low-Na + calcineurin inhibitor
With tissue from the same hearts, incubation in low-Na solution for 20 min also significantly increased Ser368-Cx43 phosphorylation (pSer368-Cx43; Fig. 2C,D; p < 0.001, n = 6). pSer368-Cx43 data are presented as normalised to total Cx43 (T-Cx43) expression. T-Cx43 expression in turn was normalised to GAPDH (1.09 ± 0.17 vs 1.09 ± 0.07 in control and low-Na solutions, n = 8; p > 0.05) and this ratio was unaffected by any interventions used: thus T-Cx43/GAPDH levels are not further reported. In concordance with the Gj data, the increase of pSer368-Cx43/T-Cx43 expression ratio in low-Na solution remained (p < 0.001, n = 6), but significantly less so in the presence of CysA (p < 0.01, n = 6). However, the increase of pSer368-Cx43/T-Cx43 expression ratio was completely prevented by CAIP.
Gap junction conductance and the role of protein phosphatases PP1 and PP2A
The PP1 inhibitor tautomycin (TTM, 5 nM) had no effect on Gj, either in control or in low-Na solution (Fig. 3A, left panel; n = 6). These data were paralleled by a lack of effect of TTM on pSer368-Cx43 expression in control or in low-Na solution (Fig. 3A, right panel; n = 6). The PP2A inhibitor fostriecin (FST, 100 nM; n = 6) also had no effect on Gj or pSer368-Cx43 levels in control solution (Fig. 3B, left and right panels. respectively). However, in low-Na solution FST partially reversed the reduction of Gj, (p < 0.01, n = 6), as well as the increase of pSer368-Cx43 expression (p < 0.001, n = 6). In all interventions T-Cx43 expression remained unchanged with respect to control.

Tautomycin (TTM) and fostriecin (FST) on gap junction conductance, Gj, and Cx43 phosphorylation in normal and low-Na solution. A Effect of TTM. Left; Gj in low-Na solution as percentage of values in control and in the absence or presence of TTM (n = 6, left panel). Right; protein expression of pSer368-Cx43 normalised to total Cx43 (T-Cx43) in control and low-Na solution in the absence or presence of TTM (n = 6, right panel). B Effect of FST on the same experimental paradigm. ***p < 0.001; low-Na vs control: §§p < 0.01; §§§p < 0.001 low-Na vs low-Na + calcineurin inhibitor
Gap junction conductance: CaMKII and PKC inhibitors
The CaMKII inhibitors KN-93 (5 µM) and AIP (10 µM) had no significant effect on Gj, either in control or in low-Na solutions (Fig. 4A). In the absence or presence of KN-93, in low-Na solution Gj was 59.3 ± 6.8 vs 63.7 ± 9.9% of control in low-Na solution (p > 0.05, n = 6). For AIP experiments, in low-Na solution Gj was 52.3 ± 8.7 vs 55.8 ± 11.0% control in its presence or absence (p > 0.05, n = 6). Conversely, the PKC inhibitor chelerythrine (2 µM) prevented the decrease of Gj in low-Na solution (Fig. 4B). In low-Na solution Gj was reduced to 42.7 ± 10.9% vs 99.0 ± 5.5% of control in the absence or presence of chelerythrine, respectively (p < 0.01, n = 5).

CaMKII and PKC inhibitors on gap junction conductance, Gj. A Effect on Gj of the CaMKII inhibitors KN93 (left) and AIP (right). In control and low-Na solution (both n = 6). B Effect on Gj of the PKC inhibitor chelerythrine (n = 5) ***p < 0.001; low-Na vs control
Phosphorylation of IP-Cx40 in low-Na solution, the calcineurin pathway
Total Cx40 protein was extracted from left atrial lysates by immunoprecipitation. Control experiments showed IP-Cx40 bound to Cx40-loaded beads, but unbound to Cx43-loaded beads. Total Cx40 (T-Cx40) was unaltered in low-Na solution (Fig. 5A, upper panels). However low-Na solution increased pSer-Cx40 and pThr-Cx40 expression. These actions of low-Na solutions were prevented by CAIP (upper panels). In addition, increase of pSer-Cx40 and pThr-Cx40 expression in low-Na solution were unaffected by TTM, but partially prevented by FST (Fig. 5A, lower panels).

Phosphorylation of serine and threonine Cx40 residues; effects of calcineurin and phosphatase inhibitors. A Representative Western blots for the detection of T-Cx40, pSer-Cx40 and pThr-Cx40 phosphorylation. Upper panel set: Lane 1, molecular weight markers; lanes 2–4, control tissue, lanes 5–10, low-Na treated tissues (lanes 8–10 also treated with CAIP). Lower panel set: Lane 1, molecular weight markers; lanes 2,3 control tissue, lanes 5–9, low-Na treated tissues (lanes 7,8 and 9,10 also treated with TTM and FST respectively). B Signal quantitation of pSer-Cx40 data normalised to T-Cx40 for control and low-Na data (in the additional presence of CysA, CAIP, FST and TTM). C: Signal quantitation of pThr-Cx40 data normalised to T-Cx40 as in part B. ***p < 0.001; low-Na vs control: §§p < 0.01; §§§p < 0.001 low-Na vs low-Na + CysA or FST
Quantitative data for expression of pSer-Cx40 and pThr-Cx40 in low-Na solution, normalised to T-Cx40 levels, show an overall picture similar to that with pSer368-Cx43. Phosphorylation of pSer-Cx40 and pThr-Cx40 was low under control conditions (Fig. 5B,C) but was significantly increased in low-Na solution ((p < 0.001, n = 6). The raised pSer-Cx40 and pThr-Cx40 expression levels were partially prevented by CysA (p < 0.001, n = 6), and completely by CAIP. TTM had no significant effect on raised pSer-Cx40 and pThr-Cx40 expression in low Na-solution, but FST partially prevented the increase of pSer-Cx40 and pThr-Cx40 in low-Na solution (p < 0.001, n = 6).
Discussion
Raised [Ca2+]i, gap junction conductance and Cx43, Cx40 phosphorylation—the role of calcineurin
This study has shown that increased [Ca2+]i in guinea-pig left atrial myocardium reduced gap junction electrical conductance (Gj) by approximately 50%; and also increased Cx43 phosphorylation at serine368 (pSer368-Cx43), as well as Cx40 phosphorylation at serine and threonine residues. These changes were fully prevented by the selective calcineurin inhibitor CAIP, and partially so by the less selective agent cyclosporin-A. Moreover, changes were partly prevented by fostriecin, but unaffected by tautomycin suggesting an intermediate regulation of calcineurin action by the phosphatase PP2A, but none from PP1.
Mode of action of Cn to regulate gap junction conductance
These observations differ in certain respects from equivalent experiments with guinea-pig ventricular myocardium. The basic observation that raised [Ca2+]i reduces Gj and this is accompanied by increased ser368-Cx43 phosphorylation by a calcineurin-dependent pathway is common to both. However, calcineurin regulates this process in different ways. In the ventricle, calcineurin dephosphorylates an inhibitor of PP1 (I1) to activate PP1 itself and initiate ser365-Cx43 dephosphorylation; this in turn enables ser368-Cx43 phosphorylation [12]. In left atrial myocardium PP1 does not have an intermediary role for the action of calcineurin, as judged by the lack of action of its inhibitor tautomycin to prevent changes when [Ca2+]i is raised. However, PP2A has at least a partial intermediary role as fostriecin partially prevented the changes to Gj and ser368-Cx43 phosphorylation induced by raising [Ca2+]I, this contrasts the lack of action of PP2A in ventricular myocardium [12]. This suggests the potential for differential regulation of gap junction electrical properties in atrial and ventricular myocardium when, for example, specific control of atrial or ventricular electrical disturbances is needed.
The additional role of Cx40 to determine Gj is a further differentiating feature of atrial myocardium. A similar increase of the phosphorylation of Cx40 serine and threonine residues was also mediated by a calcineurin-dependent pathway, with a partial role for PP2A, but none for PP1. However, it is unclear which particular phosphorylation sites on Cx40 regulate the conductance of gap junctions containing this subtype [19]. The significance of PP2A activity requires further study as this general name refers to a large family of proteins [20] and the particular subtype in guinea-pig needs to be referenced to that in human left atrium [21].. However, a number of residues that alter activity may be phosphorylated, e.g. Tyr307-PP2A that decreases enzyme activity [22], and the role of calcineurin here requires evaluation.
Another potential Ca2+-regulated route to control Gj is by phosphorylation of Ser368-Cx43 by activation of CaMKII by Ca-calmodulin. However, this was deemed unlikely in left atrium as the CaMKII inhibitors KN-93 and AIP had no significant effect on Gj in low-Na solution. However, CaMKII could have an indirect role in sarcoplasmic reticulum Ca2+ dysregulation, further maintaining high [Ca2+]i during AF [23]. By contrast, the PKC inhibitor chelerythrine prevented the reduction of Gj in low-Na solution and suggests that Cx43-Ser368 phosphorylation was mediated by PKC. A proposed scheme summarising the role of calcineurin in regulating the phosphorylation pattern of gap junction proteins is shown in Fig. 6.

Regulation of myocardial gap junction electrical conductance (Gj) by calcineurin. Gap junction (GJ) conductance is controlled by varying the phosphorylation of particular residues on connexin proteins (Cx43, Cx40) – two residues are shown (1, 2) one of which is phosphorylated (top GJ in diagram) to yield a high conductance channel. A rise of intracellular [Ca2+], [Ca2+]i, activates the phosphatase, calcineurin (Cn) to dephosphorylate residue-1 (step 1). This enables protein kinase-C (PKC) to phosphorylate residue-2, yielding a low conductance channel (step 2). Left atrial (LA) and left ventricular (LV) myocardium differ in the way Cn dephosphorylates residue-1. With LA tissue, data from this study are consistent with a direct action of Cn on residue-1, and partial intermediation of the phosphatase PP2A. With LV tissue, data (ref (12)) are consistent with dephosphorylation and deactivation of an inhibitor (I1) of the phosphatase PP1. Particular residues -1 and -2 on Cx43 are serine365 and serine368; but those on Cx40 involve unknown serine and threonine residues. The phosphatase inhibitors used are shown in italics under the relevant enzyme; see text for more details
Calcineurin-dependent pathways and atrial fibrillation (AF)
Calcineurin-dependent pathways may influence the incidence and persistence of atrial arrhythmias by several routes. Firstly, calcineurin expression is greater in human left atrial samples from patients with AF [24], which could result from increased calpain activity, the up-stream activator of Cn [25], and/or up-regulation of RCAN1 that transcribes a Cn regulator [26, 27].. Secondly, reduction of Gj, as shown in this study, will reduce the velocity of action potential propagation and contribute to emergence of re-entrant rhythms characteristic of AF. The importance of Ca2+/calcineurin-dependent pathways to determine Cx43 and Cx40 phosphorylation is an important feature. We showed previously a paradoxical age-dependent increase of Cx40 and Cx43 expression in human left atrium, despite an accompanying decrease of Gj [5].. This further underlines the importance not of connexin expression per se but the extent of post-transcriptional modification as a crucial determinant of Gj. Thirdly, calcineurin will affect ion channel current, either directly [28, 29] or by altering transcription of channel components. Calcineurin can exist either as CnAα or CnAβ, the former having rapid cytoplasmic actions, the latter acting as a chaperone for the transcription factor NFAT to enter the nucleus and initiate transcription [30, 31]. Overall calcineurin-dependent pathways can alter the electrical properties of myocardium either through long- or short-term routes: a short-term role during acute increase of intracellular [Ca2+] will importantly involve modification of gap-junction electrical properties.
Changes to gap junction conductance and impact on action potential conduction velocity (CV)
The quantitative effect of the above changes to Gj on CV in atrial and ventricular myocardium may be estimated; in this case when intracellular [Ca2+]i was raised – a high-Ca condition. This is relevant due to the pro-arrhythmic effects of reduced CV in a high-Ca state [12, 32]. CV is a positive function of total intracellular electrical conductance, (Gi=(Gj·Gc)/(Gj+Gc)) – see also Table 1. One-dimensional cable theory shows that CV = k.√(Gi), where k is a composite constant of other electrical properties of conducting myocardium [3, 33]. Thus, relative changes to Gi are determined not just by changes to Gj, but also by their proportional relation to sarcoplasmic conductance, Gc, which does not change under experimental conditions. Determination of atrial values of Gj and Gc reported here, and for ventricular myocardium in [5], give a unique opportunity to quantify more accurately the impact of altered gap junction properties on CV in these two tissues (Table 1).
The data show that gap junction conductance, Gj, is significantly greater in atrial than in ventricular myocardium in both control and high-Ca conditions, but sarcoplasmic conductance, Gc, is similar in both tissues and under control and high-Ca conditions. In consequence, total intracellular conductance, Gi, is greater in atrial myocardium in both control and high-Ca conditions. The larger value of Gi in atrial myocardium is consistent with a greater CV compared to ventricular myocardium [3]. In ventricular myocardium a reduction of Gj by about 50% (i.e. [12]) is estimated to reduce CV by about 20%, if all other biophysical characteristics [3, 33] remain constant. However, in atrial myocardium a similar proportional reduction of Gj would have a significantly smaller effect on CV, due to the greater proportional impact that Gc has on total intracellular conductance. Thus, a similar impact on atrial gap junction electrical properties would have a smaller consequence on CV, compared with ventricular myocardium.
Limitations
-
1.
Avoidance of signal drift with impedance measurements is important to compare effects of interventions with controls. Each experiment included short-term and long-term time controls: A short-term control was two ‘impedance runs’, five minutes apart for each intervention: data were not used if the difference exceeded 5%. A long-term control was inclusion of a second Tyrode’s intervention after exposure to low-Na solution. Data from preparations were also not used if Gj values differed by more than 10%: in total such drift occurred is four of 44 preparations.
-
2.
Vehicle controls were not included to minimise use of animals. In particular, stock solutions of CysA, FST and KN-93 were dissolved in DMSO, with final solvent concentrations of 0.05, 0.0005 and 0.01% (v/v) respectively. Other work has shown DMSO to affect cell membrane biophysical properties only above 0.5–1% concentration [34].
Conclusions
Calcineurin-dependent phosphorylation of connexin proteins (Cx40 and Cx43) is a key determinant of gap junction conductance and hence action potential propagation in left atrial myocardium. This particular calcineurin-dependent pathway, with the partial involvement of PP2A, is different in left atrium compared to ventricular myocardium and raises the possibility of targeted anti-arrhythmic therapies aimed more specifically at the two chambers.
Data availability
Reasonable requests for data will be considered by Prof Christopher Fry.
Abbreviations
- AF:
-
Atrial fibrillation
- AIP:
-
Autocamtide-2-Related Inhibitory Peptide, CaMKII inhibitor
- AP:
-
Action potential
- CAIP:
-
Calcineurin auto-inhibitory peptide
- Chelerythrine:
-
PKC inhibitor
- Cn:
-
Calcineurin
- Cx:
-
Connexin
- CaMKII:
-
Calcium-calmodulin dependent protein kinase II
- pSer368-Cx43:
-
Phosphorylated Cx43 at serine 368
- CysA:
-
Cyclosporin-A, Cn inhibitor
- FST:
-
Fostriecin, PP2A inhibitor
- Gc :
-
Specific sarcoplasmic (cytoplasmic) conductance
- Gi :
-
Specific total intracellular conductance
- Gj :
-
Specific gap junction conductance
- I1:
-
Inhibitor 1 (of PP1)
- IP:
-
Immunoprecipitation
- KN-93:
-
CaMKII inhibitor
- LA:
-
Left atrium
- mS:
-
MilliSiemen, unit of electrical conductance
- PKC:
-
Protein kinase C
- PP:
-
Protein phosphatase
- PP1:
-
Protein phosphatase 1
- PP2A:
-
Protein phosphatase 2A
- Ser:
-
Serine
- ‘specific’:
-
Cellular electrical constants expressed independent of cell/tissue dimensions
- Thr:
-
Threonine
- TTM:
-
Tautomycin, PP1 inhibitor
References
Link MS, Giugliano RP, Ruff CT, Scirica BM, Huikuri H, Oto A, Crompton AE, Murphy SA, Lanz H, Mercuri MF, Antman EM, Braunwald E, ENGAGE AF-TIMI 48 Investigators (2017) Stroke and mortality risk in patients with various patterns of atrial fibrillation. Circ Arrhythm Electrophysiol 10:e004267. https://doi.org/10.1161/CIRCEP.116.004267
Landstrom AP, Dobrev D, Wehrens XHT (2017) Calcium signaling and cardiac arrhythmias. Circ Res 120:1969–1993. https://doi.org/10.1161/CIRCRESAHA.117.310083
Dhillon PS, Gray R, Kojodjojo P, Jabr R, Chowdhury R, Fry CH, Peters NS (2013) Relationship between gap-junctional conductance and conduction velocity in mammalian myocardium. Circ Arrhythm Electrophysiol 6:1208–1214. https://doi.org/10.1161/CIRCEP.113.000848
Lin X, Gemel J, Glass A, Zemlin CW, Beyer EC, Veenstra RD (2010) Connexin40 and connexin43 determine gating properties of atrial gap junction channels. J Mol Cell Cardiol 48:238–245. https://doi.org/10.1016/j.yjmcc.2009.05.014
Dhillon PS, Chowdhury RA, Patel PM, Jabr R, Momin AU, Vecht J, Gray R, Shipolini A, Fry CH, Peters NS (2014) Relationship between connexin expression and gap-junction resistivity in human atrial myocardium. Circ Arrhythm Electrophysiol 7:321–329. https://doi.org/10.1161/CIRCEP.113.000606
Kanagaratnam P, Rothery S, Patel P, Severs NJ, Peters NS (2002) Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties. J Am Coll Cardiol 39:116–123. https://doi.org/10.1016/s0735-1097(01)01710-7
Lampe PD, Lau AF (2004) The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 36:1171–1186. https://doi.org/10.1016/S13572725(03)00264-4
Hood AR, Ai X, Pogwizd SM (2017) Regulation of cardiac gap junctions by protein phosphatases. J Mol Cell Cardiol 107:52–57. https://doi.org/10.1038/aps.2009.92
Huang RY, Laing JG, Kanter EM, Berthoud VM, Bao M, Rohrs HW, Townsend RR, Yamada KA (2011) Identification of CaMKII phosphorylation sites in connexin43 by high-resolution mass spectrometry. J Proteome Res 10:1098–1109. https://doi.org/10.1021/pr1008702
Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF (2000) Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol 149:1503–1512. https://doi.org/10.1083/jcb.149.7.1503
Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD (2007) Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol 179:1301–1309. https://doi.org/10.1083/jcb.200707060
Jabr RI, Hatch FS, Salvage SC, Orlowski A, Lampe PD, Fry CH (2016) Regulation of gap junction conductance by calcineurin through Cx43 phosphorylation: implications for action potential conduction. Pflügers Arch 468:1945–1955. https://doi.org/10.1007/s00424-016-1885-7
van Rijen HV, van Veen TA, Hermans MM, Jongsma HJ (2000) Human connexin40 gap junction channels are modulated by cAMP. Cardiovasc Res 45:941–951. https://doi.org/10.1016/s0008-6363(99)00373-9
Salameh A, Dhein S (2011) Adrenergic control of cardiac gap junction function and expression. Naunyn Schmiedebergs Arch Pharmacol 383:331–346. https://doi.org/10.1007/s00210-011-0603-4
Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M et al (2020) The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol 18:e3000410. https://doi.org/10.1371/journal.pbio.3000410
Cooklin M, Wallis WR, Sheridan DJ, Fry CH (1997) Changes in cell-to-cell electrical coupling associated with left ventricular hypertrophy. Circ Res 80:765–771. https://doi.org/10.1161/01.res.80.6.765
Fry CH, Salvage SC, Manazza A, Dupont E, Labeed FH, Hughes MP, Jabr RI (2012) Cytoplasm resistivity of mammalian atrial myocardium determined by dielectrophoresis and impedance methods. Biophys J 103(11):2287–2294. https://doi.org/10.1016/j.bpj.2012.10.023
Tse G, Yeo JM, Tse V, Kwan J, Sun B (2016) Gap junction inhibition by heptanol increases ventricular arrhythmogenicity by reducing conduction velocity without affecting repolarization properties or myocardial refractoriness in Langendorff-perfused mouse hearts. Mol Med Rep 14:4069–4074. https://doi.org/10.3892/mmr.2016.5738
Pogoda K, Kameritsch P, Retamal MA, Vega JL (2016) Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: a revision. BMC Cell Biol 17:11. https://doi.org/10.1186/s12860-016-0099-3
Sontag JM, Sontag E (2014) Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci 7:16. https://doi.org/10.3389/fnmol.2014.00016
Gergs U, Trapp T, Bushnaq H, Simm A, Silber RE, Neumann J (2019) Age-dependent protein expression of serine/threonine phosphatases and their inhibitors in the human cardiac atrium. Adv Med article ID 2675972. https://doi.org/10.1155/2019/2675972
Yao X, Zhang X, Yin Y, Liu B, Luo D, Liu D, Chen N, Ni Z, Wang X, Wang Q, Wang J, Liu G (2011) Glycogen synthase kinase-3β regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not Src. Biochem J 437:335–344. https://doi.org/10.1042/BJ20110347
Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnok N, Neumann K, Siepelt R, Schindube FA, Hasenfuss G, Maier LS (2010) CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 106:1134–1144. https://doi.org/10.1161/CIRCRESAHA.109.203836
Zhao Y, Cui G, Zhou N, Li C, Zhang Q, Sun H, Han B, Zou C, Wang L, Li X, Wang J (2016) Calpain-calcineurin-nuclear factor signaling and the development of atrial fibrillation in patients with valvular heart disease and diabetes. J Diab Res: Article ID 4639654. https://doi.org/10.1155/2016/4639654
Brundel BJ, Ausma J, van Gelder IC, van der Want JJ, van Gilst WH, Ctijns HJ, Henning RH (2002) Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc Res 54:380–389. https://doi.org/10.1016/s0008-6363(02)00289-4
Zhao F, Zhang S, Chen L, Wu Y, Qin J, Shao Y, Wang X, Chen Y (2012) Calcium- and integrin-binding protein-1 and calcineurin are upregulated in the right atrial myocardium of patients with atrial fibrillation. Europace 14:1726–1733. https://doi.org/10.1093/europace/eus149
Tan N, Chung MK, Smith JD, Hsu J, Serre D, Newton DW, Castel L, Soltesz E, Pettersson G, Gillinov AM, Van Wagoner DR, Barnard J (2013) Weighted gene coexpression network analysis of human left atrial tissue identifies gene modules associated with atrial fibrillation. Circ Cardiovasc Genet 6:362–371. https://doi.org/10.1161/CIRCGENETICS.113.000133
Matthes J, Jäger A, Handrock R, Groner F, Mehlhorn U, Schwinger RH, Varadi G, Schwartz A, Herzig S (2004) Ca2+-dependent modulation of single human cardiac L-type calcium channels by the calcineurin inhibitor cyclosporine. J Mol Cell Cardiol 36:241–255. https://doi.org/10.1016/j.yjmcc.2003.11.013
Qi XY, Yeh YH, **ao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S (2008) Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res 103:845–854. https://doi.org/10.1161/CIRCRESAHA.108.175463
Greenwood IA, Ledoux J, Sanguinetti A, Perrino BA, Leblanc NJ (2004) Calcineurin A-alpha but not A-beta augments ICl(Ca) in rabbit pulmonary artery smooth muscle cells. J Biol Chem 279:38830–38837. https://doi.org/10.1074/jbc.M406234200
Jabr RI, Wilson AJ, Riddervold MH, Jenkins AH, Perrino BA, Clapp LH (2007) Nuclear translocation of calcineurin A-beta but not calcineurin A-alpha by platelet-derived growth factor in rat aortic smooth muscle. Am J Physiol Cell Physiol 292:C2213-2225. https://doi.org/10.1152/ajpcell.00139.2005
Kléber AG, Rudy Y (2004) Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 84:431–488. https://doi.org/10.1152/physrev.00025.2003
Jack JJB, Noble D, Tsien RW (1976) Electric current flow in excitable cells. Oxford University Press (ISBN 13: 9780198573654)
Pan F, Mills SL, Massey SC (2007) Screening of gap junction antagonists on dye coupling in the rabbit retina. Vis Neurosci 24:609–618. https://doi.org/10.1017/S0952523807070472
Acknowledgements
The authors are very grateful to Dr Aamir Ahmed (University College London) for critically reading the final manuscript.
Funding
Research was funded by a grant from The HASTE Foundation to CHF and RIJ.
Author information
Authors and Affiliations
Contributions
Conceptualisation: RIJ, CHF. Methodology: SS and FH. Formal analysis and investigation: RIJ, SS, CHF. Writing – review and editing: RIJ, SS, CHF. All authors approved the final text.
Corresponding author
Ethics declarations
Ethical approval
Experiments were carried out using protocols approved by local Animal Welfare Ethical Review Bodies (AWERBs), under the auspices of a UK Home Office licence (70/7877 to CHF) and according to UK legislation under the Animals (Scientific Procedures) Act 1986, Amendment Regulations (SI 2012/3039). Experimental work adhered to ARRIVE guidelines. Human studies are not included in this work.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Jabr, R.I., Salvage, S.C., Hatch, F.S. et al. Calcineurin-dependent regulation of gap junction conductance and connexin phosphorylation in guinea pig left atrium. Pflugers Arch - Eur J Physiol 475, 583–593 (2023). https://doi.org/10.1007/s00424-023-02798-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-023-02798-9