
Abstract
Background
Intracranial teratoma represents a rare neoplasm, occurring predominantly during childhood. Characteristic symptoms depend on the location but are mainly hydrocephalus, visual disturbances, hypopituitarism, and diabetes insipidus. Initial diagnosis can be challenging due to similar radiological features in both teratomas and other lesions such as craniopharyngiomas. Gross total resection is recommended if feasible and associated with a good prognosis.
Case description
A 10-year-old girl presented with newly diagnosed growth retardation, fatigue, cephalgia and bilateral hemianopia. Further laboratory analysis confirmed central hypothyroidism and hypercortisolism. Cranial magnetic resonance imaging showed a cystic space-occupying lesion in the sellar and suprasellar compartment with compression of the optic chiasm without hydrocephalus present, suspicious of craniopharyngioma. Subsequently, an endonasal endoscopic transsphenoidal near-total tumor resection with decompression of the optic chiasm was performed. During postoperative recovery the patient developed transient diabetes insipidus, the bilateral hemianopia remained unchanged. The patient could be discharged in a stable condition, while hormone replacement for multiple pituitary hormone deficiency was required. Surprisingly, histopathology revealed conspicuous areas of skin with formation of hairs and squamous epithelia, compatible with a mature teratoma.
Conclusions
We present an extremely rare case of pediatric sellar teratoma originating from the pituitary gland and a review of literature focusing on the variation in presentation and treatment. Sellar teratomas are often mistaken for craniopharyngioma due to their similar radiographic appearances. However, the primary goal of treatment for both pathologies is to decompress eloquent surrounding structures such as the optic tract, and if applicable, resolution of hydrocephalus while avoiding damage to the pituitary stalk and especially the hypothalamic structures. If feasible, the aim of surgery should be gross total resection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intracranial germ cell tumors (ICGCT) are rare and account for approximately 3% of all central nervous system (CNS) tumors in children and young adults and they rarely occur after the 4th decade of life [1,2,3]. The incidence of pediatric ICGCTs varies considerably between different regions. The incidence in Europe and the United States is around 0.5–3% of all CNS tumors, while in Asia, especially Japan, the incidence ranges up to 8–14% [4,5,6]. The genetic factors influencing this difference are not fully elucidated [7].
ICGCTs mostly arise in the midline, either in the sellar or pineal region. Based on the WHO classification of CNS tumors, intracranial germ cell tumors comprise a heterogeneous group of neoplasms, divided into germinomas and non-germinomatous germ cell tumors (NGGCT) [1, 4, 8]. While germinomas account for up to 50–70% of ICGCTs, NGGCTs are further subdivided into embryonal carcinomas, yolk sac tumors, choriocarcinomas, mature teratomas, immature teratomas, and mixed germ cell tumors [4, 10]. Mature teratomas consist of fully differentiated elements derived from one or all three germ cell layers, while immature teratoma contains incompletely differentiated tissue [9]. Despite the fact that teratoma may manifest anywhere in the midline, most occur in the pineal region and only few cases of pediatric sellar teratomas are described in the literature [6, 10,11,12,13,14,15,16,17,18,19,20].
In this article we present a case report of a ten-year-old girl presenting with a suprasellar mature teratoma and provide a literature review thereof focusing on the treatment options.
Case description
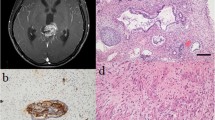
A ten-year-old girl with a history of cystic fibrosis (CF) with gastrointestinal and pulmonary involvement, presented to her pediatrician with newly diagnosed growth retardation, fatigue and frequent headache. An arginine growth hormone-releasing hormone test confirmed growth hormone (GH) deficiency, and after further laboratory analysis, a central hypothyroidism and hypercortisolism was diagnosed. Substitutional therapy with levothyroxine and hydrocortisone was initiated, which improved the patient’s headache and fatigue. Ophthalmologic examination revealed bitemporal hemianopia. Further work-up with magnetic resonance imaging (MRI) revealed a 3 × 3 × 2.5 cm cystic space-occupying lesion in the sellar and suprasellar compartment with compression of the optic chiasm, with partial calcifications in computed tomography (CT), highly suspicious of a craniopharyngioma (CP) (Fig. 1A–C). Hydrocephalus was not present at the time of presentation. Due to the compression of the chiasm causing clinical hemianopia, the indication for surgical decompression was given. We weighed the possibilities of a neuroendoscopic transventricular (NET) cyst fenestration and partial tumor resection versus an endonasal endoscopic approach (EEA) or an open transcranial approach. The advantage for the transventricular neuroendoscopic approach is its minimal-invasive nature and straight-forward decompression of the cyst, which is causing compression of the optic chiasm. However, the patient presented with very small ventricles, making neuroendoscopy cumbersome, and the neuroendoscopic approach would only allow a partial tumor resection. EEA shares the advantage of being minimal-invasive. However, due to her age and the concomitant diagnosis of CF, known to be associated with hypoplasia and markedly reduced pneumatization of the paranasal sinuses, a non-pneumatized (conchal type) sphenoid was present (Fig. 1D), making the AAE more challenging [21]. In addition, gross total resection (GTR) was not considered achievable through an EEA, due to a supra-chiasmatic tumor extension. Still, both an NET or EEA seemed superior to an open approach (e.g. subfrontal or interhemispheric) given the invasiveness and associated morbidity of such approaches, while similar to the EEA and NET approach, GTR would most probably not be achieved either [22,23,24,25,26]. We ultimately decided to perform an EEA together with our colleagues from ENT. The conchal configuration of the sphenoid sinus required meticulous drilling of squamous intrasphenoidal bone, exposure of the harder sellar bone, and a superior trans-chiasmatic sulcus extension to achieve satisfactory exposure of the suprasellar tumor cyst (Fig. 2). Intraoperatively, crystals and cystic fluid, suspicious of CP, were drained from the cyst, and the cyst was dissected from the cavernous sinus walls, the sellar diaphragm, and the dorsum sellae without risking injury of adjacent structures. At the end of the operation, a symmetrical diaphragmal descent was achieved as indirect sign for the decompression of the optic chiasm (Fig. 3, Supplementary Video 1). Postoperative MRI showed the expected near total tumor removal. While the cyst was completely drained, tumor remnants extending posteriorly to the superiorly displaced chiasm remained as expected (Fig. 4). Postoperatively, the patient developed diabetes insipidus (DI) for which she received desmopressin under the supervision of the pediatric endocrinologists. During her inpatient stay, she recovered from her DI with stable sodium levels but required vasopressin substitution. Overall recovery was good, while the hemianopia persisted. No signs of rhinorrhea resulting from cerebrospinal fluid (CSF) fistula were noted. We were able to discharge the patient to her home 11 days after surgery. Unexpectedly, the histopathologic analysis found conspicuous areas of skin with formation of hairs and squamous epithelia, compatible with a mature teratoma (Fig. 5). The cytokeratin staining was positive for epithelial cells consistent with the finding in a mature teratoma (Fig. 5B, C). These findings led to the diagnosis of a rare case of infantile mature teratoma originating from the sellar region After discussion in our interdisciplinary pediatric neuro-tumor-board, no further treatments (e.g. chemotherapy, radiation therapy) were indicated and a clinical and radiological follow up was initiated.

A Coronal T1-weighted magnetic resonance imaging (MRI). B Sagittal soft tissue-weighted computed tomography (CT). C Sagittal T1-weighted MRI demonstrating a 3 × 3 × 2.5 cm cystic space-occupying partially calcified lesion in the sellar and suprasellar compartment with compression of the optic chiasm, highly susceptive of a craniopharyngioma (CP). D Axial bone-weighted CT demonstrating a non-pneumatized sphenoid bone with a hypo intense bone-marrow (asterisk)

Endonasal endoscopic approach in a conchal type sphenoid sinus for a sellar and suprasellar cystic lesion. A Binostril approach and exposure of the rostrum sphenoidale [RS]. B Removal of the spongious and fatty sphenoidal bone marrow [M]. C Change of the color and consistency indicates exposition of the harder sellar bone [S]. D Trans-sellar exposure of the sellar dura [D]. E Trans-chiasmatic sulcus superior extension. F Complete osteodural trans-sellar trans-chiasmatic sulcus exposure prior dural opening

Endonasal endoscopic approach in a conchal type sphenoid sinus for a sellar and suprasellar cystic lesion. A Extrusion of crystals [*] and brownish “oily” cystic fluid. B Close-up inspection of the cystic content reveals solid component consisting of crystals [*] and debris. C Dissection of the suprasellar tumor cyst from the partially descending sellar diaphragm [D]. D Dissection of the tumor cyst from the left medial cavernous sinus wall [CS]. E Incomplete diaphragmal [D] descent indicating remaining suprasellar tumor components. Dissection from the left medial cavernous sinus wall [lCS] and the dorsum sellae [DS] was possible, while the cyst was very adherent to the right medial cavernous sinus wall [rCS]. F Symmetrical diaphragmal [D] descent at the end of the surgery prior skull base defect closure

Radiological course. A Preoperative sagittal and B coronal T2-weighted MRI of the cystic sellar and suprasellar infantile mature teratoma. C Immediate postoperative sagittal and D coronal T2-weighted MRI demonstrating skull base defect reconstruction material within the sphenoid cavity and the sella turcica [*], along with a decompressed and anatomically located optic chiasm and remnant solid tumor supero-posteriorly [**]. E 1-year postoperative sagittal and F coronal T2-weighted MRI demonstrating resorption of the intrasellar reconstruction material along with volumetric regression and descent of the solid tumor remnant along the pituitary stalk into the dorsal aspect of the sella turcica. The optic nerves and chiasm descent into the sellar region and remains decompressed and anatomically preserved

Histopathology of the sellar tumor. A Hematoxylin-Eosin (HE) stain 10× magnified showing areas of skin formation, circle indicates the area which is B zoomed into with a magnification of 20x. C Pan-Cytokeratin (CK22) stain, 5× magnified, proving epithelial cell presence and circle indicates the area which is D zoomed into with a magnification of 20×
At her first postoperative follow-up appointment after 6 weeks the patient was back in school, without any complaints. The hemianopia remained unchanged, and cortisol, vasopressin and thyroxin substitution was still required one year postoperatively. She was also started on growth hormone (GH) replacement therapy 6 months postoperatively. MRI follow-up one year after surgery showed stable appearances without any sign of progression (Fig. 4).
Discussion
Although intracranial teratomas account for only about 0.5% of all intracranial tumors, they represent a highly relevant differential diagnosis for pediatric midline tumors [27]. The literature on pediatric sellar teratomas is scarce, and to our knowledge only 4 cases of mature sellar teratoma have been reported to date (Table 1) [6, 10,11,12,13,14,15,16,17,18,19,20].
Symptoms are often unspecific and include headaches and signs of increased intracranial pressure in case of hydrocephalus, while visual impairment, mainly bitemporal hemianopia, panhypopituitarism and DI can be found at a more chronical stage of the disease [12, 27, 28]. In our case, multiple pituitary hormone deficiencies were diagnosed due to short stature with delayed skeletal age. A more thorough testing revealed central hypothyroidism and hypocortisolism, for which substitution therapy was initiated preoperatively. According to Esfahani et al., pediatric suprasellar germ cell tumors (GCT) usually present primarily with DI, while other symptoms due to compression of the anterior pituitary are hypocortisolism and hypothyroidism [12, 27]. The symptom complex of frequent headaches, bitemporal hemianopia, growth retardation, and DI strongly suggests the differential diagnosis of a sellar tumor, which is most commonly a CP but could be any other midline pathology in this region [30, 31]. Based on the MRI findings in our case, which showed a partially cystic and partially solid lesion (Fig. 1), the diagnosis of a CP was further substantiated. It has been previously reported that NGGCT in the sellar region can mimic CP due to their similar clinical and radiographic characteristics [15, 31,32,33]. Both tumors have radiographic hallmarks of mixed density, usually with cystic and solid components, while for mature teratomas an occasional finding of teeth, fat, hair, or calcifications is possible [17, 20, 27, 34,35,36,37]. Classically, mature teratomas contain cells of all ecto-, meso- and endodermal layers, however, not all teratomas exhibit all three tissue layers, which can make their diagnosis more challenging. Teratoma arise from the midline structures and as in most intracranial GCT they tend to occur in either the pineal region or more rarely suprasellar [38, 39]. In some cases teeth or fat tissue can be visualized on a preoperative scan, which can facilitate diagnosis, however, in most reported cases, the teratoma mimicked either a CP or a Rathke’s cleft cyst [16, 38].
Craniopharyngiomas on the other hand, arise from the Rathke’s cleft epithelium, and hence also occur in the midline. They are observed to have a bimodal distribution occurring in children and in people in their 5-6th decades of life, while teratomas mainly occur in children and young adults [40].
Intracranial NGGCTs can show elevated serum and CSF markers like alpha-fetoprotein (AFP), human beta-chorionic gonadotropin (ß-HCG) and placental alkaline phosphatase (PLAP), however, they are not typically elevated in mature teratomas [29, 41,42,43]. In our case, tumor markers were not measured, as we suspected a CP in the first place. Therefore, the diagnosis was ultimately achieved by histopathological analysis. Huang et al. recently reported that CCND2, RB1, and PRDM14 are involved in the cellular and molecular pathogenesis of IGCTs. Moreover, they suggest KIT/RAS and AKT1/mTOR signaling pathways as possible therapeutic targets [44].
In most cases of space-occupying sellar tumors with neurological deficits surgical therapy is indicated to decompress the nervous structures but also to reach an ultimate histopathological diagnosis [27, 41, 45].
Tumors originating from the sella can be reached either endoscopically through an EEA, NET, or by open pterional, interhemispheric, transfrontal-transcortical with or without a transchoroidal window, or subfrontal craniotomy, depending on the tumor extension. In young children, the transsphenoidal approach can be more challenging due to incomplete pneumatization of the sphenoid sinus, which does not usually begin until after the third year of life and is only fully developed at about 15 years of age, increasing the likelihood of intraoperative hemorrhage [22]. In addition, transsphenoidal approaches are limited for suprachiasmatic tumor extension, or lateral tumor extension, given the proximity to the carotid arteries [23, 24]. Moreover, a study showed that CSF fistulas occurred at a higher rate in EEA compared to open transcranial approaches for pediatric sellar lesions [24]. For tailored extended EEA approaches with GTR or near-total resection strategies, experience in dealing with graded repair for large skull base defects, harboring a relevant risk for high-flow CSF-leaks, is mandatory and should only be performed by experienced skull base neurosurgeons and ENT surgeons in an interdisciplinary setup [46, 47]. As our patient did not show any intraoperative CSF flow, along with a symmetrical descent of the diaphragm, a defect reconstruction using only resorbable hemostyptic materials without the need of an abdominal fat graft or pedicled vascularized flap was justified (Fig. 3,Video 1). Overall, EEA is appropriate for resection of pediatric tumors of the sellar region and shows good results, if done in experienced centers and interdisciplinary manner including pediatric neurosurgeons, skull-base surgeons and ENT surgeons [25, 48]. Another endoscopic approach is the NET for tumor cyst fenestration via the 3rd ventricle, which is also a minimal invasive method, however an extensive resection of the tumor can hardly be achieved due to limited hemostatic control and access route. Open surgery is the traditional approach and feasible for large tumors, yet the localization of the optic nerves, the carotid arteries and the hypothalamus must be carefully considered preoperatively. Further, open resection carries additional risk of morbidity due to brain retraction potentially causing seizures, hemorrhage, or ischemia [24]. In all of these approaches, it is of paramount importance that the hypothalamus is not injured during surgery, and in case of hypothalamic tumor invasion, GTR should not be the main aim of surgery but rather an intended near-total resection, as in our case (Fig. 3,Video 1) [25, 49]. In children, hypothalamic injury results in hyperphagia, obesity, neurocognitive deficits, and lower quality of life and should be avoided at all costs, especially before puberty, independent of the underlying diagnosis [50,51,52]. Other common complications after sellar/suprasellar surgery include diabetes insipidus (DI), endocrine disturbances or CSF leak [36, 53, 54]. Our patient suffered from postoperative DI, requiring long-term treatment with desmopressin. A recent meta-analysis by Lee et al. concluded that approximately one quarter of all patients developed new permanent DI after surgery for pediatric sellar and suprasellar lesions [54].
Children diagnosed with GH deficiency need to be closely monitored to avoid any disproportionate growth or growth retardation. According to a recent consensus statement on the safety of hormone replacement in survivors of cancer and intracranial including pituitary tumors, the timing of initiation of GH therapy after surgery is multifactorial and should be decided individually and discussed thoroughly with all parties involved.
Radiological evidence of tumor stability is always a prerequisite for start of therapy. However, in children with radiologically stable craniopharyngiomas showing significant growth and metabolic disturbances, this may be considered as early as 3 months after the end of tumor therapy. Furthermore, based on current scientific evidence, there appears to be no association between GH replacement and increased cancer-related mortality in childhood cancer survivors [55]. Our patient did not only suffer from hormonal deficiency but also had a history of CF. While the literature often mentions an association between CF and various types of cancer, mainly gastrointestinal tumors, but also skin, lung, hematological and breast cancers, there is no evidence of an association between CF and brain tumors or cystic teratomas in general [56,57,58,59,60,61,62,63,64,65,66,67,68].
The recurrence rate of mature teratoma after GTR is low and long-term outcome is excellent with a reported 10-year survival rate of 93% [27, 69, 70]. Hence, GTR is considered the gold standard treatment and mature teratomas represent the only entity of germ cell tumors that can be treated by resection alone [27, 69, 71]. Other germ cell tumors such as immature teratomas or mixed GCT teratomas, require a multistage treatment involving surgical resection, adjuvant or neoadjuvant chemo- and radiotherapy [27, 39, 70]. In case of postoperative tumor remnant of a mature teratoma, like in our case, current treatment recommendation is radiographic observation and in case of a progression either re-resection, if surgically feasible, or radiotherapy [72]. Some reports suggest that Gamma knife radiosurgery may even be considered superior to conventional radiotherapy in the recurrence treatment of mature teratomas minimizing the risk of pituitary dysfunction as well as of hypothalamic damage [73,74,75]. Regardless of the extent of resection, especially pediatric patients will be under radiographic surveillance for numerous years and often require life-long hormone substitution. The importance to follow these patients over years and the difficulty of continuing care for these patients during transition to adulthood has been discussed within the literature [76,77,78,79,80,81,82,83,84].
Conclusion
To our knowledge, this is one of very few case reports describing a pediatric sellar mature teratoma. Initial diagnosis can be challenging due to similar radiological features with CP, which is more frequent in this age group. In case the tumor causes compression of the optic chiasm or hydrocephalus, surgery is warranted regardless of the diagnosis. Depending on the tumor extension (suprasellar, third ventricle, lateral of the carotids) the most suitable surgical approach should be chosen. Moreover, when considering EEA in younger children, the pneumatization patterns of the paranasal sinuses and adjacent skull base structures should be analyzed carefully, and only an experienced interdisciplinary team including pediatric and skull base neurosurgeons, ENT surgeons and pediatric endocrinologists should treat these patients. Mature teratomas have a good overall prognosis, however these children require long-term follow up within the setting of a pediatric interdisciplinary neuro-oncological clinic.
References
Kong Z, Wang Y, Dai C, Yao Y, Ma W, Wang Y (2018) Central nervous system germ cell tumors: A review of the literature. J Child Neurol 33(9):610–620. https://doi.org/10.1177/0883073818772470
Hoffman HJ et al (1991) Intracranial germ-cell tumors in children. J Neurosurg 74(4):545–551. https://doi.org/10.3171/jns.1991.74.4.0545
Juliano J, Melamed E, Christian E, Tamrazi B, Krieger MD (2019) Imaging features predictive of recurrence in pediatric intracranial germ-cell tumors. Pediatr Neurosurg 54(3):173–180. https://doi.org/10.1159/000493194
Echevarría ME, Fangusaro J, Goldman S (2008) Pediatric central nervous system germ cell tumors: a review. Oncologist 13(6):690–699. https://doi.org/10.1634/theoncologist.2008-0037
Lee SH et al (2017) Nationwide population-based incidence and survival rates of malignant central nervous system germ cell tumors in Korea, 2005–2012. Cancer Res Treat 49(2):494–501. https://doi.org/10.4143/crt.2016.129
Tsoukalas N et al (2013) Coexistence of intracranial germ cell tumor and craniopharyngioma in an adolescent: case report and review of the literature. Int J Clin Exp Med 6(3):211–218
Takami H et al (2021) Comparison on epidemiology, tumor location, histology, and prognosis of intracranial germ cell tumors between Mayo Clinic and Japanese consortium cohorts. J Neurosurg 134(2):446–456. https://doi.org/10.3171/2019.11.JNS191576
Wu C-C et al (2017) MRI features of pediatric intracranial germ cell tumor subtypes. J Neurooncol 134(1):221–230. https://doi.org/10.1007/s11060-017-2513-x
Louis DN et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol 23(8):1231–1251. https://doi.org/10.1093/neuonc/noab106
Fults D, Kelly DL (1983) A suprasellar atypical teratoma presenting as an intrasellar mass: a case report. Neurosurgery 13(1):40–43. https://doi.org/10.1227/00006123-198307000-00007
Lee Y-J, Lin JCT, Shen E-Y, Liang D-C, Wong T-T, Huang F-Y (1995) Loss of visibility of the neurohypophysis as a sign of central diabetes insipidus. Eur J Radiol 21(2):145–147. https://doi.org/10.1016/0720-048X(95)00700-Z
Cho DY, Wang YC, Ho WL (1997) Primary intrasellar mixed germ-cell tumor with precocious puberty and diabetes insipidus. Childs Nerv Syst 13(1):42–46. https://doi.org/10.1007/s003810050038
Ikeda J, Sawamura Y, Kato T, Abe H (1998) Metachronous neurohypophyseal germinoma occurring 8 years after total resection of pineal mature teratoma. Surg Neurol 49(2):205–208. https://doi.org/10.1016/s0090-3019(97)00158-4
Yagi K, Kageji T, Nagahiro S, Horiguchi H (2004) Growing teratoma syndrome in a patient with a non-germinomatous germ cell tumor in the neurohypophysis-case report-. Neurol Med Chir (Tokyo) 44(1):33–37. https://doi.org/10.2176/nmc.44.33
Chang CV, Nunes VD, Felicio AC, Zanini MA, Cunha-Neto MB, Castro AV (2008) Mixed germ cell tumor of the pituitary-hypothalamic region presenting as craniopharyngioma: case report and review of the literature. Arq Bras Endocrinol Metabol 52(9):1501–1504. https://doi.org/10.1590/S0004-27302008000900015
Vendrell J-F, Hoa D, Gahide G (2010) Mature teratoma arising from the sella. Lancet 375(9725):1556. https://doi.org/10.1016/S0140-6736(09)60300-7
Kim YS, Kang SG, Kim YO (2010) Pituitary teratoma presenting as central diabetes insipidus with a normal MRI finding. Yonsei Med J 51(2):293–294. https://doi.org/10.3349/ymj.2010.51.2.293
Chiloiro S, Giampietro A, De Marinis L (2015) Safety of rhGH replacement therapy in suprasellar pituitary teratoma: A Case-Report. Int J Endocrinol Metab Disord 1(2). https://doi.org/10.16966/2380-548X.107
Yoneoka Y, Yoshimura J, Sano M, Okada M, Kakita A, Fujii Y (2018) Third ventricle germ cell tumor originating from the infundibulum with rapidly expansive enlargement. Pediatr Neurosurg 53(1):49–54. https://doi.org/10.1159/000480021
Araki K, Kola M, Okada T, Kurashige T, Naruse K, Hiroi M (2000) A boy with normal growth in spite of growth hormone deficiency after resection of a suprasellar teratoma. Endocr J 47(SupplMarch):S101–S104. https://doi.org/10.1507/endocrj.47.SupplMarch_S101
Marino MJ, Riley CA, Wu EL, Weinstein JE, McCoul ED (2020) The unified airway: Does asthma influence paranasal sinus pneumatization? Ear Nose Throat J 99(2):89–93. https://doi.org/10.1177/0145561319848992
Oviedo P, Levy ML, Nation J (2020) Approaching the sella through the nonpneumatized sphenoid in pediatric patients. J Neurol Surg Part B Skull Base 81(1):56–61. https://doi.org/10.1055/s-0039-1679895
Elliott RE, Jane JA Jr, Wisoff JH (2011) Surgical management of craniopharyngiomas in children: meta-analysis and comparison of transcranial and transsphenoidal approaches. Neurosurgery 69(3):630–643. https://doi.org/10.1227/NEU.0b013e31821a872d
Komotar RJ, Starke RM, Raper DMS, Anand VK, Schwartz TH (2012) Endoscopic endonasal compared with microscopic transsphenoidal and open transcranial resection of craniopharyngiomas. World Neurosurg 77(2):329–341. https://doi.org/10.1016/j.wneu.2011.07.011
Alalade AF et al (2018) Suprasellar and recurrent pediatric craniopharyngiomas: expanding indications for the extended endoscopic transsphenoidal approach. J Neurosurg Pediatr 21(1):72–80. https://doi.org/10.3171/2017.7.PEDS17295
Silva AHD, Aquilina K (2019) Surgical approaches in pediatric neuro-oncology. Cancer Metastasis Rev 38(4):723–747. https://doi.org/10.1007/s10555-019-09832-2
Chiloiro S, Giampietro A, Bianchi A, De Marinis L (2016) Clinical management of teratoma, a rare hypothalamic-pituitary neoplasia. Endocrine 53(3):636–642. https://doi.org/10.1007/s12020-015-0814-4
Nishioka H, Ito H, Haraoka J, Akada K (1999) Immature teratoma originating from the pituitary gland: case report. Neurosurgery 44(3):644–647. https://doi.org/10.1097/00006123-199903000-00115
Esfahani DR, Alden T, DiPatri A, ** G, Goldman S, Tomita T (2020) Pediatric suprasellar germ cell tumors: a clinical and radiographic review of solitary vs. bifocal tumors and its therapeutic implications. Cancers 12(9):2621. https://doi.org/10.3390/cancers12092621
Petito CK, DeGirolami U, Earle KM (1976) Craniopharyngiomas: a clinical and pathological review. Cancer 37(4):1944–1952. https://doi.org/10.1002/1097-0142(197604)37:4%3c1944::aid-cncr2820370446%3e3.0.co;2-#
Zada G, Lin N, Ojerholm E, Ramkissoon S, Laws ER (2010) Craniopharyngioma and other cystic epithelial lesions of the sellar region: a review of clinical, imaging, and histopathological relationships. Neurosurg Focus 28(4):E4. https://doi.org/10.3171/2010.2.FOCUS09318
Youl BD, Plant GT, Stevens JM, Kendall BE, Symon L, Crockard HA (1990) Three cases of craniopharyngioma showing optic tract hypersignal on MRI. Neurology 40(9):1416–1419. https://doi.org/10.1212/wnl.40.9.1416
Nada R, Mohan H, Dhir SP, Mukherjee KK, Kak VK (2000) Suprasellar malignant mixed germ cell tumour presenting as craniopharyngioma. Neurol India 48(4):381–384
Choi SH, Kwon BJ, Na DG, Kim J-H, Han MH, Chang K-H (2007) Pituitary adenoma, craniopharyngioma, and Rathke cleft cyst involving both intrasellar and suprasellar regions: differentiation using MRI. Clin Radiol 62(5):453–462. https://doi.org/10.1016/j.crad.2006.12.001
Johnson LN, Hepler RS, Yee RD, Frazee JG, Simons KB (1986) Magnetic resonance imaging of craniopharyngioma. Am J Ophthalmol 102(2):242–244. https://doi.org/10.1016/0002-9394(86)90152-2
Bogusz A, Müller HL (2018) Childhood-onset craniopharyngioma: latest insights into pathology, diagnostics, treatment, and follow-up. Expert Rev Neurother 18(10):793–806. https://doi.org/10.1080/14737175.2018.1528874
Chapman PR, Singhal A, Gaddamanugu S, Prattipati V (2020) Neuroimaging of the pituitary gland: practical anatomy and pathology. Radiol Clin North Am 58(6):1115–1133. https://doi.org/10.1016/j.rcl.2020.07.009
Jiang S, Wang Z, You Y, Wang R, Bao X (2021) Suprasellar mature cystic teratoma mimicking rathke’s cleft cyst: a case report and systematic review of the literature. Front Endocrinol 12:731088. https://doi.org/10.3389/fendo.2021.731088
Jennings MT, Gelman R, Hochberg F (1985) Intracranial germ-cell tumors: natural history and pathogenesis. J Neurosurg 63(2):155–167. https://doi.org/10.3171/jns.1985.63.2.0155
Kobalka PJ, Huntoon K, Becker AP (2021) Neuropathology of pituitary adenomas and sellar lesions. Neurosurgery 88(5):900–918. https://doi.org/10.1093/neuros/nyaa548
Murray MJ, Bartels U, Nishikawa R, Fangusaro J, Matsutani M, Nicholson JC (2015) Consensus on the management of intracranial germ-cell tumours. Lancet Oncol 16(9):e470–e477. https://doi.org/10.1016/S1470-2045(15)00244-2
Haase J, Børgaard-Pedersen B (1979) Alpha-feto-protein (AFP) and human chorionic gonadotropin (HCG) as biochemical markers of intracranial germ-cell tumours. Acta Neurochir (Wien) 50(1–2):67–69. https://doi.org/10.1007/BF01813551
Allen JC, Nisselbaum J, Epstein F, Rosen G, Schwartz MK (1979) Alphafetoprotein and human chorionic gonadotropin determination in cerebrospinal fluid: an aid to the diagnosis and management of intracranial germ-cell tumors. J Neurosurg 51(3):368–374. https://doi.org/10.3171/jns.1979.51.3.0368
Huang X, Zhang R, Mao Y, Zhou L-F, Zhang C (2016) Recent advances in molecular biology and treatment strategies for intracranial germ cell tumors. World J Pediatr 12(3):275–282. https://doi.org/10.1007/s12519-016-0021-2
Murray MJ, Horan G, Lowis S, Nicholson JC (2013) Highlights from the Third International Central Nervous System Germ Cell Tumour symposium: laying the foundations for future consensus. Ecancermedicalscience 7:333. https://doi.org/10.3332/ecancer.2013.333
Roethlisberger M, Govindaraju R, Rychen J, Jadczak A, Brand Y, Jeyaganesh V, Mun KS, Hench J, Mariani L, Prepageran N (2021) Chapter 1. Pathologies of the Sellar Region. In: Narayanan J, Prepageran N (eds) Atlas of 360 degree skull base surgery, 1st edn. Thieme Medical and Scientific Publishers Private Limited, pp 3–93
Gutierrez WR, Bennion DM, Walsh JE, Owen SR (2020) Vascular pedicled flaps for skull base defect reconstruction. Laryngoscope Investig Otolaryngol 5(6):1029–1038. https://doi.org/10.1002/lio2.471
Kuan EC et al (2019) Lack of sphenoid pneumatization does not affect endoscopic endonasal pediatric skull base surgery outcomes. Laryngoscope 129(4):832–836. https://doi.org/10.1002/lary.27600
Puget S et al (2007) Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg 106(1 Suppl):3–12. https://doi.org/10.3171/ped.2007.106.1.3
Puget S (2012) Treatment strategies in childhood craniopharyngioma. Front Endocrinol 3:64. https://doi.org/10.3389/fendo.2012.00064
Bogusz A, Boekhoff S, Warmuth-Metz M, Calaminus G, Eveslage M, Müller HL (2019) Posterior hypothalamus-sparing surgery improves outcome after childhood craniopharyngioma. Endocr Connect 8(5):481–492. https://doi.org/10.1530/EC-19-0074
Fouda MA, Karsten M, Staffa SJ, Scott RM, Marcus KJ, Baird LC (2021) Management strategies for recurrent pediatric craniopharyngioma: new recommendations. J Neurosurg Pediatr 1–8. https://doi.org/10.3171/2020.9.PEDS20606
Wu D et al (2022) Extended neuroendoscopic endonasal approach for resection of craniopharyngioma in children. Front Neurol 13:771236. https://doi.org/10.3389/fneur.2022.771236
Lee JA, Cooper RL, Nguyen SA, Schlosser RJ, Gudis DA (2020) Endonasal endoscopic surgery for pediatric sellar and suprasellar lesions: a systematic review and meta-analysis. Otolaryngol Head Neck Surg 163(2):284–292. https://doi.org/10.1177/0194599820913637
Boguszewski MCS et al (2022) Safety of growth hormone replacement in survivors of cancer and intracranial and pituitary tumours: a consensus statement. Eur J Endocrinol 186(6):P35–P52. https://doi.org/10.1530/EJE-21-1186
Sheldon CD, Hodson ME, Carpenter LM, Swerdlow AJ (1993) A cohort study of cystic fibrosis and malignancy. Br J Cancer 68(5):1025–1028. https://doi.org/10.1038/bjc.1993.474
Garg M, Ooi CY (2017) The enigmatic gut in cystic fibrosis: linking inflammation, dysbiosis, and the increased risk of malignancy. Curr Gastroenterol Rep 19(2):6. https://doi.org/10.1007/s11894-017-0546-0
Slae M, Wilschanski M (2021) Cystic fibrosis and the gut. Frontline Gastroenterol 12(7):622–628. https://doi.org/10.1136/flgastro-2020-101610
Scott P, Anderson K, Singhania M, Cormier R (2020) Cystic fibrosis, CFTR, and colorectal cancer. Int J Mol Sci 21(8):2891. https://doi.org/10.3390/ijms21082891
Anderson KJ, Cormier RT, Scott PM (2019) Role of ion channels in gastrointestinal cancer. World J Gastroenterol 25(38):5732–5772. https://doi.org/10.3748/wjg.v25.i38.5732
Hough NE, Chapman SJ, Flight WG (2020) Gastrointestinal malignancy in cystic fibrosis. Paediatr Respir Rev 35:90–92. https://doi.org/10.1016/j.prrv.2020.03.002
Archangelidi O et al (2022) Incidence and risk factors of cancer in individuals with cystic fibrosis in the UK: a case-control study. J Cyst Fibros Off J Eur Cyst Fibros Soc 21(2):302–308. https://doi.org/10.1016/j.jcf.2021.07.004
Maisonneuve P, Lowenfels AB (2022) Cancer in cystic fibrosis: a narrative review of prevalence, risk factors, screening, and treatment challenges: adult cystic fibrosis series. Chest 161(2):356–364. https://doi.org/10.1016/j.chest.2021.09.003
Appelt D, Fuchs T, Steinkamp G, Ellemunter H (2022) Malignancies in patients with cystic fibrosis: a case series. J Med Case Reports 16(1):27. https://doi.org/10.1186/s13256-021-03234-1
Rousset-Jablonski C et al (2022) Cancer incidence and prevalence in cystic fibrosis patients with and without a lung transplant in France. Front Public Health 10:1043691. https://doi.org/10.3389/fpubh.2022.1043691
Appelt D, Steinkamp G, Ellemunter H (2022) Cancer in cystic fibrosis: do not neglect gynecologic cancers. Chest 161(5):e325–e326. https://doi.org/10.1016/j.chest.2022.01.055
FitzMaurice TS, Nazareth DS (2022) Incidence of breast cancer in people with cystic fibrosis: A cause for concern? J Cyst Fibros Off J Eur Cyst Fibros Soc 21(5):890. https://doi.org/10.1016/j.jcf.2021.11.017
Shi X et al (2022) Integrative pan cancer analysis reveals the importance of CFTR in lung adenocarcinoma prognosis. Genomics 114(2):110279. https://doi.org/10.1016/j.ygeno.2022.110279
Nishio S, Inamura T, Takeshita I, Fukui M, Kamikaseda K (1993) Germ cell tumor in the hypothalamo-neurohypophysial region: clinical features and treatment. Neurosurg Rev 16(3):221–227. https://doi.org/10.1007/BF00304332
Matsutani M et al (1997) Primary intracranial germ cell tumors: a clinical analysis of 153 histologically verified cases. J Neurosurg 86(3):446–455. https://doi.org/10.3171/jns.1997.86.3.0446
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Craniopharyngioma Treatment. Bethesda, MD: National Cancer Institute. Available at: https://www.cancer.gov/types/brain/hp/child-cranio-treatment-pdq. Accessed 21 Jan 2024. PMID: 26389330
Aoyama H et al (1998) Retrospective multi-institutional study of radiotherapy for intracranial non-germinomatous germ cell tumors. Radiother Oncol 49(1):55–59. https://doi.org/10.1016/S0167-8140(98)00081-4
Chiu C-D, Chung W-Y, Pan DH-C, Wong T-T, Shih Y-H, Lee L-S (2006) Gamma knife radiosurgery for intracranial mature teratoma–long-term results and review of literature. Surg Neurol 65(4):343–351. https://doi.org/10.1016/j.surneu.2005.07.075
Hasegawa T, Kondziolka D, Hadjipanayis CG, Flickinger JC, Lunsford LD (2003) Stereotactic radiosurgery for CNS nongerminomatous germ cell tumors. Report of four cases. Pediatr Neurosurg 38(6):329–333. https://doi.org/10.1159/000070417
Kobayashi T, Kida Y, Mori Y (2001) Stereotactic gamma radiosurgery for pineal and related tumors. J Neurooncol 54(3):301–309. https://doi.org/10.1023/a:1012727306066
Ebel F, Greuter L, Guzman R, Soleman J (2022) Transitional care in pediatric brain tumor patients: a systematic literature review. Child Basel Switz 9(4):501. https://doi.org/10.3390/children9040501
Tallen G et al (2015) Strategies to improve the quality of survival for childhood brain tumour survivors. Eur J Paediatr Neurol 19(6):619–639. https://doi.org/10.1016/j.ejpn.2015.07.011
Campbell F et al (2016) Transition of care for adolescents from paediatric services to adult health services. Cochrane Database Syst Rev 4:CD009794. https://doi.org/10.1002/14651858.CD009794.pub2
Roux A et al (2021) Toward a transitional care from childhood and adolescence to adulthood in surgical neurooncology? A lesson from the Necker-Enfants Malades and the Sainte-Anne Hospitals collaboration. J Neurosurg Pediatr 28(4):380–386. https://doi.org/10.3171/2021.3.PEDS2141
Vinchon M, Dhellemmes P (2007) The transition from child to adult in neurosurgery. Adv Tech Stand Neurosurg 32:3–24. https://doi.org/10.1007/978-3-211-47423-5_1
Eshelman-Kent D, Gilger E, Gallagher M (2009) Transitioning survivors of central nervous system tumors: challenges for patients, families, and health care providers. J Pediatr Oncol Nurs 26(5):280–294. https://doi.org/10.1177/1043454209343209
Heitzer AM, Ris D, Raghubar K, Kahalley LS, Hilliard ME, Gragert M (2020) Facilitating transitions to adulthood in pediatric brain tumor patients: the role of neuropsychology. Curr Oncol Rep 22(10):102. https://doi.org/10.1007/s11912-020-00963-2
Janss AJ, Mazewski C, Patterson B (2019) Guidelines for treatment and monitoring of adult survivors of pediatric brain tumors. Curr Treat Options Oncol 20(1):10. https://doi.org/10.1007/s11864-019-0602-0
Nicklin E et al (2021) Unmet support needs in teenage and young adult childhood brain tumour survivors and their caregivers: ‘it’s all the aftermath, and then you’re forgotten about.’ Support Care Cancer 29(11):6315–6324. https://doi.org/10.1007/s00520-021-06193-x
Funding
Open access funding provided by University of Basel No funding was received for this study.
Author information
Authors and Affiliations
Contributions
Conceptualization: JS, RG. Methodology/Review: KK, LG. Case Description: KK, LG, MR, SF. Writing - original draft preparation: KK, LG. Writing - review and editing: all authors. Supervision: JS, RG.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Supplementary Video 1 Intra- and suprasellar cystic-solid infantile mature teratoma in a child with conchal type sphenoid sinus – Extended endonasal endoscopic transsphenoidal trans-sellar and chiasmatic-sulcus approach, subtotal tumor resection and skull base defect reconstruction (MP4 548083 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kürner, K., Greuter, L., Roethlisberger, M. et al. Pediatric sellar teratoma – Case report and review of the literature. Childs Nerv Syst 40, 1259–1270 (2024). https://doi.org/10.1007/s00381-024-06296-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-024-06296-w