
Abstract
The spike protein determines the host-range specificity of coronaviruses. In particular, the Receptor-Binding Motif in the spike protein from SARS-CoV-2 contains the amino acids involved in molecular recognition of the host Angiotensin Converting Enzyme 2. Therefore, to understand how SARS-CoV-2 acquired its capacity to infect humans it is necessary to reconstruct the evolution of this important motif. Early during the pandemic, it was proposed that the SARS-CoV-2 Receptor-Binding Domain was acquired via recombination with a pangolin infecting coronavirus. This proposal was challenged by an alternative explanation that suggested that the Receptor-Binding Domain from SARS-CoV-2 did not originated via recombination with a coronavirus from a pangolin. Instead, this alternative hypothesis proposed that the Receptor-Binding Motif from the bat coronavirus RaTG13, was acquired via recombination with an unidentified coronavirus. And as a consequence of this event, the Receptor-Binding Domain from the pangolin coronavirus appeared as phylogenetically closer to SARS-CoV-2. Recently, the genomes from coronaviruses from Cambodia (bat_RShST182/200) and Laos (BANAL-20-52/103/247) which are closely related to SARS-CoV-2 were reported. However, no detailed analysis of the evolution of the Receptor-Binding Motif from these coronaviruses was reported. Here we revisit the evolution of the Receptor-Binding Domain and Motif in the light of the novel coronavirus genome sequences. Specifically, we wanted to test whether the above coronaviruses from Cambodia and Laos were the source of the Receptor-Binding Domain from RaTG13. We found that the Receptor-Binding Motif from these coronaviruses is phylogenetically closer to SARS-CoV-2 than to RaTG13. Therefore, the source of the Receptor-Binding Domain from RaTG13 is still unidentified. In accordance with previous studies, our results are consistent with the hypothesis that the Receptor-Binding Motif from SARS-CoV-2 evolved by vertical inheritance from a bat-infecting population of coronaviruses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
How the coronavirus SARS-CoV-2 evolved to infect humans continues to be an active area of research. To understand the origin of the zoonosis it is crucial to identify the closest viral wild population from which SARS-CoV-2 originated. Very early during the pandemic, the bat coronavirus RaTG13 from China’s Yunnan province, was identified as the most closely related to SARS-CoV-2, showing an average genome-wide nucleotide identity of 96.1% (Zhou et al. 2020).
This was followed by the proposal that the Receptor-Binding Domain (RBD) of the spike protein from SARS-CoV-2 was acquired by recombination with pangolin-infecting coronaviruses (Li et al. 3). In both cases, the Bayes factor (BCS) is larger than 100 which is interpreted as decisive evidence against the SEPARATE hypothesis (Kass and Raftery 1995). Therefore, the evolution of the gene coding for the spike protein for these 14 coronaviruses is better described by 4 recombination breakpoints.
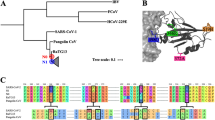
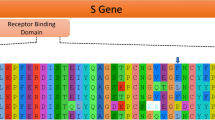
Next, we focused on the phylogenetic history of the concatenated segments 2 and 4 versus the segment 3. Segments 2 and 4 contain a small fraction of the NTD, the RBD (minus the RBM), SD1, SD2, FP and a fragment of the IFP motif (Fig. 3); while segment 3 corresponds to the RBM. The topology of the phylogenetic trees of these segments are almost identical except for three exceptions (Fig. 4). In the first place, the coronavirus Guangxi_P4L is in a different bipartition in the tree inferred from segment 3 (the tree in the right of Fig. 4), although the posterior probability of the internal node supporting this bipartition is low (0.61), raising doubts on its veracity. Second, the coronaviruses Guangdong 1 and bat_RShSTT182 interchange positions between the two trees, but again, the posterior probability supporting the position of Guangdong 1 is low (0.58) in the tree inferred from segment 3. And most importantly, in the segment 3 tree (Fig. 4 right), the coronavirus RaTG13 branches outside the well supported bipartition (0.91) defined by the coronaviruses: bat_RShSTT182, Guangdong 1, Wuhan-Hu-1/2019, BANAL-20-103 and BANAL-20-52, thus supporting the hypothesis that the RBM in RaTG13 was acquired via recombination with a yet unknown coronavirus (Boni et al. 2020).
We further evaluated the phylogenetic dissonance (D) between the two trees in Fig. 4 by using GALAX software. Dissonance is a measure of phylogenetic conflict between segments/partitions of data; and is estimated by measuring the average information content in Bayesian posterior tree samples from individual segments minus the information contained in the merged set of Bayesian tree samples from all segments (Lewis et al. 2016). D takes values from 0 to 1 (or 0 to 100%) where 0 indicates no phylogenetic conflict between segments. Dissonance between two trees can be further partitioned by clades. This is, it is possible to identify which clades contribute most to dissonance between trees.
In Fig. 4 we show which clades contribute most to dissonance (D) between trees. The largest percentage to dissonance (43%) is contributed by the partition that divides the tree between the external group (Rco319) and the rest of the OTUs. This is expected because the two trees are different as a whole. However, the second percentage to dissonance is contributed by the partition containing coronaviruses most closely related to Wuhan-Hu-1/2019, including RaTG13 (69%–43% = 26%), these are depicted with red doted lines connecting the two trees. On the third place, is the contribution to dissonance of the partition that includes the above species plus GuanxiP4L and RSYN04 (56%–43% = 13%), these are depicted with orange lines. This result further reinforces that segment 3 has a different phylogenetic history than segments 2 and 4. The coverage of the dissonance analysis is 0.78 (see supplementary material for a complete description of the statistics associated with the dissonance analysis).
The origin of the RBM by recombination in RaTG13 was further confirmed by analysis with the Recombination Detection Program (RDP) (Martin et al. 2020). This software applies several different methodologies to a set of sequences and calculates an overall consensus score to assess the veracity of detected recombination events. A full exploratory recombination scan identified a recombination event with high confidence (consensus score > 60) between RaTG13 and an unknown coronavirus at positions 1308 to 1514 of the multiple sequence alignment. These coordinates correspond to the RBM and coincides with that detected by GARD (see supplementary material).
The next question is whether the immediate co-descendant to the clade conformed by Wuhan-Hu-1/2019, BANAL-20-52 and BANAL-20-103 is the bat (bat_RShST188) or the pangolin (Guangdong 1) coronavirus. This is important because it would indicate if the RBM from Wuhan-Hu-1/2019 descend from a coronavirus that infects bats or pangolins.
Given that the posterior probability of the node supporting the close relationship of Guangdong 1 to the clade containing Wuhan-Hu-1/2019, BANAL-20-52 and BANAL-20-103 in the tree inferred from segment 3 is low (0.58; Fig. 4, right), one possibility is that the closest coronavirus to the clade containing Wuhan-Hu-1/2019 is bat_RShST188, as shown in the tree inferred from segments 2 and 4 in Fig. 4 (left). In fact, an alternative phylogeny to that shown in Fig. 4 (right) were Guangdong 1 shifts position with bat_RShST188, is not significatively worse than the original tree according to a Kishino-Hasegawa test (p value = 0.341) (see supplementary material). Therefore, the hypothesis that the RBM from SARS-CoV-2 evolved from a bat infecting coronavirus cannot be rejected.
If we use this alternative topology where Guangdong 1 shifts position with bat_RShST188 to reconstruct the ancestral sequences, we find that the RBM of the common ancestor of Wuhan-Hu-1/2019 and Guangdong 1 (the sequence named as “ancestral 4” in Fig. 5) was identical to that of Wuhan-Hu-1/2019, with the exception of residue Q498H. Showing that natural selection did not favor changes in the RBM of these coronaviruses to adapt to new hosts since they last shared a common ancestor. The same result is obtained if the original tree (the one shown in Fig. 4 right) is used for the ancestral sequence reconstruction (see supplementary material).

Ancestral sequence reconstruction shows that the RBM of the common ancestor of Wuhan-Hu-1/2019, BANAL-20-52, BANAL-20-103, bat_RShST188 and Guangdong 1 (here named as “ancestral_4”) was identical to the RBM of Wuhan-Hu-1/2019, except for the residue Q498H (red arrow). Amino acids involved in human ACE2 recognition are indicated with arrows
Discussion
The analyses provided here shows that the RBM from RaTG13 is not closely related to the RBM from SARS-CoV-2 and was likely acquired by recombination with a yet unknown coronavirus (Boni et al. 2020). Because of that, the RBM from the coronaviruses from Laos (BANAL-20-52 and BANAL-20-103) are the most closely related to the RBM from SARS-CoV-2 (Temmam et al. 2022).
Our results also show that SARS-CoV-2 did not acquire its RBM by recombining with a pangolin infecting coronavirus. Instead, our analyses indicate that the coronaviruses Wuhan-Hu-1/2019, BANAL-20-52 and BANAL-20-103 inherited its RBM most likely from a bat infecting coronavirus. Parsimony favors this interpretation given that bat_RShST188, Wuhan-Hu-1/2019, BANAL-20-52 and BANAL-20-103 are all bat-infecting coronaviruses. If recombination between a bat and pangolin infecting coronaviruses played a role in the evolution of the RBM (or the whole RBD), this may have occurred prior to the divergence of bat_RShST188 and Wuhan-Hu-1/2019.
Our results are in agreement with the interpretation of Temmam et al. (2022) regarding the evolution of the RBM in SARS-CoV-2. Accordingly, natural selection did not incidentally improve the affinity of the RBM for human ACE2 in an intermediate host before spillover (Makarenkov et al. 2021), nor did selection optimize the RBM in humans early after spillover (Andersen et al. 2020). This follows from the fact that the RBM from SARS-CoV-2 is identical to the ancestral sequence it shared with Guangdong 1 with the exception of a single amino acid change Q498H. The conservation of the RBM between coronaviruses that infects pangolins, bats and humans is consistent with recent research showing that SARS-CoV-2 is a generalist virus that is not specifically adapted to humans (Li et al. 2023). However, the origin(s) of other peculiarities of SARS-CoV-2, like the furin-cleavage site, remain to be elucidated. Such features may have evolved by different mechanisms that may have included the passage of the coronavirus in an intermediate host.
Material and Methods
Gene sequences from the spike protein were retrieved from the GenBank and GISAID databases (https://gisaid.org/; Khare et al. 2021). Author acknowledgments for sequences downloaded from GISAID are provided in supplementary material. The spike protein coding genes were extracted from genome sequences following annotation. When annotation was not available, we identified the spike coding gene by aligning the gene from Wuhan-Hu-1/2019 to the query genome using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Codon multiple sequence alignment was performed in MEGA software 11v (Tamura et al. 2021).
Recombination analysis was done with GARD as implemented in http://datamonkey.org/ (Weaver et al. 2018) with the following parameters: normal run mode, universal genetic code, without site-to-site rate variation and 2 rate classes.
Domains in the spike protein follow those defined by Lan et al. (2020) and ** stone algorithm as implemented in MrBayes (Ronquist et al. 2012). Next, the Bayes factor BF is:
These authors suggest the following interpretation of BF:
log10(BCS) | BCS | Evidence against MS |
|---|---|---|
0 to ½ | 1 to 3.2 | Not worth more than a bare mention |
½ to 1 | 3.2 to 10 | Substantial |
1 to 2 | 10 to 100 | Strong |
> 2 | > 100 | Decisive |
Phylogenetic dissonance, D, was calculated with GALAX software (https://github.com/plewis/galax). To generate the sample trees required for GALAX, we ran the mcmc algorithm in MrBayes with 1,000,000 generations and a sampling frequency of 500. The model was set to: GTR + G + I. Phylogenetic trees from Fig. 4 were inferred with MrBayes with the same parameters and 25% of burnin was discarded from the sample. For step**-stone analysis the ss algorithm was set to run 1,000,000 generations and sample each 1000 generation. Example files to run mcmc and ss algorithms in MrBayes are provided in supplementary material.
A full exploratory recombination scan was applied to the multiple sequence alignment with the program RDP (Martin et al. 2020). Methods used within RDP were: RDP, GENECONV, BootScan, MaxChi, Chimera, SiScan and 3Seq. Default parameters were used and sequences were assumed to be linear. We further asked RDP to save a distributed alignment with recombinant regions separated. Based on this distributed alignment we inferred a Maximum-Likelihood tree with MEGA11 (100 bootstrap replicas and GTR + G model of sequence evolution). For clarity, we included in this tree only the recombinant sequence corresponding to the RBM from RaTG13.
Figure 3 was generated with Circos (Krzywinski et al. 2009). Kishino-Hasegawa test was implemented in IQ-TREE (Kishino and Hasegawa 1989; Minh et al. 2020). Ancestral sequence reconstruction (ASR) was performed in MEGA software 11v by Maximum-Likelihood under the Tamura-3 parameter model and including all sites (Tamura et al. 2021). Multiple sequence alignment was visualized with Jalview (Waterhouse et al. 2009).
Supplementary material.
Change history
25 June 2024
A Correction to this paper has been published: https://doi.org/10.1007/s00239-024-10183-y
References
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452. https://doi.org/10.1038/s41591-020-0820-9
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Ta N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533. https://doi.org/10.1093/nar/gkq399
Ben Chorin A, Masrati G, Kessel A, Narunsky A, Sprinzak J, Lahav S, Ashkenazy H, Ben-Tal N (2020) ConSurf-DB: an accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci 29:258–267. https://doi.org/10.1002/pro.3779
Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW et al (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiol 5:1408–1417. https://doi.org/10.1038/s41564-020-0771-4
Cui J, Li F, Shi Z-L (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17(3):181–192. https://doi.org/10.1038/s41579-018-0118-9
Delaune D, Hul V, Karlsson EA, Hassanin A, Ou TP et al (2021) A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nature Commun 12(1):6563. https://doi.org/10.1038/s41467-021-26809-4
Kass RE, Raftery AE (1995) Bayes factors. J Am Stat Assoc 90:773–795. https://doi.org/10.2307/2291091
Khare S, Gurry C, Freitas L, Schultz MB, Bach G et al (2021) GISAID’s role in pandemic response. China CDC Weekly 3(49):1049–1051. https://doi.org/10.46234/ccdcw2021.255
Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J Mol Evol 29:170–179. https://doi.org/10.1007/BF02100115
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW (2006) Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 23(10):1891–1901. https://doi.org/10.1093/molbev/msl051
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645. https://doi.org/10.1101/gr.092759.109
Lam TTY, Jia N, Zhang YW, Shum MH-H, Jiang J-F et al (2020) Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583:282–285. https://doi.org/10.1038/s41586-020-2169-0
Lan J, Ge J, Yu J, Shan S, Zhou H et al (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220. https://doi.org/10.1038/s41586-020-2180-5
Lewis PO, Chen M-H, Kuo L, Lewis LA, Fučíková K et al (2016) Estimating Bayesian phylogenetic information content. Syst Biol 65(6):1009–1023. https://doi.org/10.1093/sysbio/syw042
Li X, Giorgi EE, Marichannegowda MH, Foley B, **ao Ch et al (2020) Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv. https://doi.org/10.1126/sciadv.abb9153
Li M, Du J, Liu W, Li Z, Lv F et al (2023) Comparative susceptibility of SARS-CoV-2, SARS-CoV, and MERS-CoV across mammals. ISME J 17:549–560. https://doi.org/10.1038/s41396-023-01368-2
Lytras S, Hughes J, Martin D, Swanepoel P et al (2022) Exploring the natural origins of SARS-CoV-2 in the light of recombination. Genome Biol Evol 14(2):evac018. https://doi.org/10.1093/gbe/evac018
Makarenkov V, Mazoure B, Rabusseau G, Legrendre P (2021) Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin. BMC Ecol Evol 1:5. https://doi.org/10.1186/s12862-020-01732-2
Martin DP, Varsani A, Roumagnac P, Botha G, Maslamoney S, Schwab T, Kelz Z, Kumar V, Murrell B (2020) RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol 7(1):veaa087. https://doi.org/10.1093/ve/veaa087
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD et al (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol and Evol 37(5):1530–1534. https://doi.org/10.1093/molbev/msaa015
Neupane S et al (2019) Assessing combinability of phylogenomic data using Bayes factors. Syst Biol 68(5):744–754. https://doi.org/10.1093/sysbio/syz007
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM et al (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A et al (2012) MRBAYES 3.2: efficient Bayesian phylogenetic inference and model selection across a large model space. Syst Biol 61(3):539–542
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38(7):3022–3027. https://doi.org/10.1093/molbev/msab120
Temmam S, Vongphayloth K, Baquero E, Munier S, Bonomi M et al (2022) Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 604:330–336. https://doi.org/10.1038/s41586-022-04532-4
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Pond SLK (2018) Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol 35(3):773–777. https://doi.org/10.1093/molbev/msx335
**a X (2021) Domains and functions of spike protein in SARS-Cov-2 in the context of vaccine design. Viruses 13(1):109. https://doi.org/10.3390/v13010109
Zhang T, Wu Q, Zhang Z (2020) Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol 30(7):1346-1351.e2. https://doi.org/10.1016/j.cub.2020.03.022
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. https://doi.org/10.1038/s41586-020-2012-7
Acknowledgements
We gratefully acknowledge all data contributors, i.e. the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. This work was supported by a CONAHCyT postdoctoral research fellowship “Estancias Posdoctorales por México” (l1200/320/2022, l1200/224/2021) awarded to Lizbeth Román-Padilla. Finally, we want to thank the students that took the course on Molecular Evolution (May, 2023) at Cinvestav Irapuato with whom we discussed a previous version of this manuscript. This work was funded by Consejo Nacional de Humanidades, Ciencias y Tecnologías de México (Conahcyt grants FORDECYT-PRONACES/103000/2020 and CB-2016-01/284992).
Author information
Authors and Affiliations
Contributions
Both authors contributed to the study conception and design. LRP designed the Bayesian factor and dissonance analysis and LD performed the molecular evolution analyses. The first draft version of the manuscript was written by LD and LRP commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing financial interest.
Additional information
Handling editor: Keith Crandall.
The original online version of this article was revised: The figure 4 was revised.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Delaye, L., Román-Padilla, L. Untangling the Evolution of the Receptor-Binding Motif of SARS-CoV-2. J Mol Evol 92, 329–337 (2024). https://doi.org/10.1007/s00239-024-10175-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-024-10175-y