
Abstract
Since many years, acrylamide (AA) is a well-known toxicologically relevant processing contaminant (“food-borne toxicant”). However, only during the recent years, high levels of acrylamide have been reported in vegetable chips. In the present study, AA was quantitated via a modified derivatization procedure with 2-mercaptobenzoic acid based on stable isotope dilution analysis and liquid chromatography–mass spectrometry. Extraction with a modified QuEChERS (quick, easy, cheap, efficient, rugged, safe) method, defatting with n-hexane, and a solid phase extraction clean-up with strong cation-exchange material were performed prior to the derivatization step. Limits of detection and quantitation (LoD and LoQ) were 12 and 41 µg of AA/kg of vegetable chips (estimated via signal-to-noise ratios of 3:1 and 10:1, respectively), and thus below the LoQ of 50 µg/kg requested by the European Food Safety Authority. Recovery rates between 92 and 101% at four spiking levels with a good precision expressed as a relative standard deviation < 7% were determined. With this method at hand, a survey of the current AA amounts in 38 vegetable chips from the worldwide market was performed, showing a remarkable variability between the different vegetables, but also between different products of the same vegetable. Thereby, the AA amounts ranged between 77.3 and 3090 µg/kg, with an average of 954 µg/kg which was distinctly higher in comparison to commercially available potato chips also analyzed in the present study (12 samples, range: 117–832 µg/kg, average: 449 µg/kg). While for sweet potato and parsnip relatively low AA amounts were found, beetroot and carrot showed rather high contents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The “food-borne toxicant” acrylamide (AA) is a processing contaminant present in several heat-processed foods like potato chips, French fries, crispbread, and breakfast cereals [1,2,3]. AA is mainly formed during the Maillard reaction at temperatures above 120 °C in thermally processed (e.g., baked or fried) potato- and cereal-based products rich in its precursor, the free amino acid asparagine, while at the same time also the desired aroma-active compounds are formed and the color is developed [4,5,6,7].
In 1994, the International Agency for Research on Cancer (IARC) classified AA as “probably carcinogenic to humans” (Group 2A) [8]. AA was found to cause neurotoxic and carcinogenic effects in animals [9, 10] and a possible relation between dietary intake of AA and the risk of colorectal cancer in humans was presumed [11]. On the basis of monitoring data from the European Food Safety Authority (EFSA) collected during 2007 and 2008, the European Commission (EC) set indicative values for eight food product categories known to contain AA, for example, 1000 μg of AA/kg for potato chips [12]. In 2015, EFSA published a scientific opinion, indicating AA in food may increase the risk of cancer for humans [13]. Two years later, the EC set benchmark levels for eight food categories in Regulation (EC) No. 2017/2158 (enforced April 11th, 2018), for example, 500 µg of AA/kg for French fries and 750 µg of AA/kg for potato chips [14]. Therein, also measures for the reduction of AA levels for the food industry and recommendations for consumers were outlined. Further, mitigation efforts and the adjustment of the manufacturing processes and control steps have to be performed when these benchmark levels are exceeded. Manufacturers must analyze the AA levels in their products and document the applied mitigation strategies.
It was shown that especially the quantities of free asparagine and reducing sugars and the temperature and time during heat processing have major impact on the formation of AA [2, 15, 16]. Huge efforts on mitigation strategies, performed by the food industry for > 15 years, resulted in lower AA levels in potato- and cereal-based foods. For potato chips, for example, modifications in the production process and the use of potato varieties with low amounts of free asparagine and sugars were implemented [17].
In recent years, vegetable chips were designed as a ‘healthy’ alternative to potato chips and other salty snacks and gained increasing popularity. These chips mainly comprise beetroots, parsnips, sweet potatoes, and carrots and are fried in the same way as potato chips. Up to now, only a few data about the AA amounts in vegetable chips have been reported [18,19,20,21]. A very recent study showed that 50% of the samples exceeded the benchmark level for potato chips [22]. Also, Breitling-Utzmann and Hankele [23] reported on AA present in nearly all commercial vegetable chips in their study with amounts ranging from < limit of detection (LoD; 10 μg of AA/kg) up to 2100 μg of AA/kg and a median of 800 μg of AA/kg. Thereby, savoy cabbage or kale chips revealed AA amounts < 100 μg/kg, while beetroot, parsnip, carrot, and sweet potato chips contained significantly higher amounts (240–2100 μg of AA/kg). However, vegetable chips have not been included in the current list of foods for which benchmark levels exist [14]. Considering the situation, the EC recently recommended also monitoring the presence of AA in certain food categories such as vegetable chips by the Commission Recommendation (EC) No. 2019/1888 [24], why reliable data for AA levels in vegetable chips are of great interest.
In literature, numerous methods for the analysis of AA in various food categories are available and were fundamentally reviewed [25,26,27,28]. Predominantly, gas chromatography and liquid chromatography (GC and LC) both coupled with mass spectrometry (MS) were used, as methods for the underivatized analyte and various derivatized forms were accessible [27], and stable isotope dilution analysis (SIDA) was often applied [2, 7, 22, 29, 30]. Very recently, some highly sophisticated methods based on liquid chromatography–tandem mass spectrometry (LC–MS/MS) without derivatization were published including some new analytical approaches like the chromatographic separation on a hydrophilic interaction liquid chromatography (HILIC) column [31] or the extraction based on supercritical fluid chromatography that offered an eleven times higher sensitivity compared to ultrahigh-performance liquid chromatography (UHPLC) and an improvement in data acquisition time (60 samples per 12 h) [32]. A further interesting development was the slightly modified method originally published by Gökmen et al. [33] based on a single-step extraction followed by protein precipitation (Carrez) enabling a simultaneous determination of AA and 5-(hydroxymethyl)furfural using positive atmospheric pressure chemical ionization (APCI+) mode [34]. In 2003, Jezussek and Schieberle [29] developed a quantitation method for AA based on a cheaper instrumentation setup (LC–MS) after derivatization with 2-mercaptobenzoic acid leading to a conversion of AA into a stable thioether to increase the selectivity and sensitivity. Regarding the analysis of AA in vegetable chips, LC–MS methods for the determination of the underivatized analyte [18, 22, 35] and GC–MS methods for the derivatized analyte [19,20,21] have been published.
The aim of this study was the development of a reliable method for the quantitation of AA in vegetable chips after derivatization based on SIDA enabling the analysis by a cheap and unsophisticated LC–MS instrument setup. Therefore, the method of Jezussek and Schieberle [29] was modified to determine the current AA amounts in vegetable chips from the global market and compare those to the AA contents in potato chips bought in German supermarkets.
Materials and methods
Chemicals and materials
The following chemicals were commercially available: acrylamide (AA, ≥ 99%) (Merck, Darmstadt, Germany), magnesium sulfate (MgSO4, 99%) (AppliChem, Darmstadt), sodium chloride (NaCl, ≥ 99%) and sodium sulfate (anhydrous, ≥ 99%) (Carl Roth, Karlsruhe, Germany), ethanol (≥ 99.5%), lead(II) acetate (≥ 99.5%), and sodium hydroxide solution (NaOH, 1 mol/L, analytical grade) (Th. Geyer, Renningen, Germany), and acetonitrile (LC–MS grade), dichloromethane (≥ 99.9%), formic acid (LC–MS grade), n-hexane (≥ 95%, for pesticide residue analysis), hydrochloric acid (∼37%, for trace analysis), and 2-mercaptobenzoic acid (97%) (Sigma-Aldrich, Steinheim, Germany). Ultrapure water (> 18 MΩ·cm) was supplied by a Synergy system (Merck Millipore, Darmstadt).
The stable isotopically labeled internal standard (ISTD) [13C3]-acrylamide ([13C3]-AA; 99 atom % 13C, 98%) was obtained from Sigma-Aldrich.
Solid phase extraction (SPE) cartridges (OASIS MCX; 60 µm, 6 cc, 500 mg) were from Waters (Eschborn, Germany). A reversed phase (RP) 18 MS column (100 mm × 3 mm, 2.6 µm) and the corresponding pre-column (10 mm × 3 mm, 2.6 µm) were obtained from Thermo Fisher Scientific (Dreieich, Germany).
Vegetable chips (between July and December 2019) and potato chips (January 2020) were purchased from local supermarkets and from the worldwide market (via online suppliers).
Determination of method sensitivity, precision, and recovery rate
The AA stock solution (250 µg/mL) was prepared in a mixture of water/ethanol (1/1, v/v) and the stable isotopically labeled ISTD [13C3]-AA stock solution (500 µg/mL) was prepared in a mixture of water/methanol (1/1, v/v).
The limit of quantitation (LoQ) in the reagent blank was determined with an AA working solution [5 µg/mL of water/ethanol (1/1, v/v)] according to the DIN 32645 calibration graph method [36]. Therefore, mixtures consisting of the AA working solution (1.5–15 µL) and the ISTD stock solution (generally 10 µL) were derivatized as described below, leading to calibration standards of the resulting AA thioether of 15–150 µg of AA/L for LC–MS analysis. For vegetable chips, the LoD (3:1) and LoQ (10:1) were estimated via signal-to-noise (S/N) ratios of representative samples.
Calibration standards for recovery experiments were prepared as mentioned above using mixtures of the AA stock solution (1–48 µL) and the ISTD stock solution (generally 10 µL), resulting in concentrations of 0.5–24 mg of AA/L (corresponding to 83.3–4000 µg of AA/kg of sample) and of generally 10 mg of [13C3]-AA/L (both analyzed as thioether).
Sample extraction
Extraction of AA was performed by a slightly modified QuEChERS (quick, easy, cheap, efficient, rugged, safe) method according to [37]. In brief, crushed chips (3 g) were mixed with water (10 mL) in a polypropylene (PP) centrifuge tube (50 mL). The ISTD stock solution (10 µL), acetonitrile (10 mL), and n-hexane (5 mL) were added, and the sample was homogenized with an Ultra-Turrax (Janke & Kunkel; IKA, Staufen im Breisgau, Germany) at a speed of 9000 rpm for 2 min at room temperature (RT). A mixture of MgSO4 (4 g) and NaCl (0.5 g) was added, and the tube was immediately shaken vigorously at 2200 rpm for 1 min at RT using a small shaking device (VXR basic Vibrax; IKA). Then, the mixture was centrifuged at 5000 rpm for 2 min at 21 °C (Heraeus Mulitfuge X1R; Thermo Fisher Scientific), the n-hexane layer was discarded, and an aliquot of the acetonitrile phase (5 mL) was subjected to the SPE clean-up procedure.
Spiked samples for recovery experiments were prepared by adding the AA stock solution (1.2, 6, 18, and 36 µL) to a parsnip chips sample (3 g each), corresponding to 100, 500, 1500, and 3000 µg of AA/kg, prior to the sample extraction described above.
Solid phase extraction (SPE)
For SPE, MCX cartridges were equilibrated with acetonitrile (2 mL). For clean-up, an aliquot of the acetonitrile phase of the sample extract (5 mL) was transferred onto the MCX cartridge and passed through (approx. 1 mL/min; without applying vacuum but using nitrogen if needed), followed by the addition of acetonitrile (2 mL) for the complete elution of AA and [13C3]-AA. The solvent was evaporated by means of a gentle stream of nitrogen at RT and the residue was subjected to derivatization.
Derivatization
Derivatization was performed according to Jezussek and Schieberle [29] with some modifications. Standards and sample residues were dissolved in water (40 mL) and the pH value was adjusted to 9 ± 0.3 by the addition of NaOH (1 mol/L). Then, the derivatization reagent solution (1 mL; 15.4 mg of 2-mercaptobenzoic acid/mL of NaOH (1 mol/L); freshly prepared before use) was added and the reaction mixture was gently stirred in the dark for 3 h at RT. The pH value was checked every 30 min and, if necessary, re-adjusted to 9 ± 0.3. An excess of the derivatization reagent was removed by the addition of saturated lead(II) acetate solution (1.5 mL) and the mixture was centrifuged in a PP tube (50 mL) at 8000 rpm for 15 min at 21 °C (Heraeus Mulitfuge X1R). The supernatant was adjusted to a pH value of 1.5 ± 0.3 with HCl (5 mol/L) before liquid-liquid extraction (LLE) with dichloromethane (3 × 30 mL) was performed in a PP tube (50 mL) at 2200 rpm for 10 min at RT using a small shaking device (VXR basic Vibrax). The organic phases were combined, dried over anhydrous Na2SO4, and the solvent was evaporated (at ~ 680 mbar and 40 °C) to a volume of ~ 1 mL that was blown down by a stream of nitrogen at RT. The residue was dissolved in acetonitrile (300 µL), transferred into an amber glass vial (1.5 mL), and the solvent was evaporated again with a gentle stream of nitrogen at RT. Finally, the standards and sample residues were dissolved in a mixture of 0.1% aqueous formic acid/acetonitrile (7/3, v/v; 500 µL for the standards and 250 µL for the samples). The solutions were centrifuged at 13,000 rpm for 5 min at RT (Heraeus Biofuge pico; Thermo Fisher Scientific) and aliquots of the supernatants (200 µL) were used for LC–MS analysis.
High-performance liquid chromatography–mass spectrometry (HPLC–MS)
The HPLC–MS equipment consisted of an 1100 HPLC system with a quaternary pump, a vacuum solvent degasser, a column oven, and an autosampler, coupled with a G6120 single quadrupole mass spectrometer equipped with an electrospray ionization (ESI) interface (Agilent Technologies, Waldbronn, Germany). Chromatography was performed on an RP18 MS column with the corresponding pre-column at 40 °C and a flow rate of 0.4 mL/min. The injection volume was generally 5 µL, mobile phase A was 0.1% aqueous formic acid, and mobile phase B was acetonitrile. Gradient elution started with 10% B (0–0.5 min), linearly increased to 95% B (0.5–8 min), kept constant at 95% (8–12 min), decreased to 10% B (12–12.2 min), and kept constant at 10% (12.2–17 min) to equilibrate the column. The mass spectrometer was operated with the following parameters in ESI+ mode: capillary voltage 4.0 kV, skimmer voltage 35 V, lens voltage 2.5 V, quadrupole temperature 100 °C, drying gas temperature 350 °C, drying gas flow rate 10 mL/min, and nebulizer gas pressure 40 psig. To evaluate co-extracted matrix during method development, total ion current chromatograms in the scan mode were recorded from m/z 100 to 400 using the following parameters: fragmentor voltage 100 V, gain 5, threshold 100, and step size 0.10. Quantitation of AA was performed in selected ion monitoring (SIM) mode at m/z 226 for the AA thioether and m/z 229 for the [13C3]-AA thioether, using a time window of 4.5–7 min, a fragmentor voltage of 100 V, and a gain of 5. The system was operated by OpenLAB CDS ChemStation C.01.09 software (Agilent Technologies).
Sample analysis
Vegetable chips samples consisted of 4 different packages of sweet potatoes, 1 package of beetroots, and 11 packages of vegetable mixtures (assorted and measured separately). In this way, 10 samples of sweet potatoes, 10 samples of parsnips, 11 samples of beetroots, and 7 samples of carrots were obtained. For comparison, 12 different packages of potato chips were additionally investigated.
Prior to sample workup, the (assorted) vegetable chips from one complete package were crushed in PP bags. The following sample preparation, derivatization, and LC–MS analysis were performed as described above (n = 2–4) (Fig. S1 in Supplementary material). A sample extract without the addition of the ISTD was prepared for each sample as a blank (n = 1). For quantitation, the same calibration range that has been used for the recovery experiments (83.3–4000 µg of AA/kg of vegetable chips) was applied. The concentration of the ISTD was generally 10 mg/L. Quantitation was performed via the peak areas of the AA thioether and the [13C3]-AA thioether in combination with the amounts of ISTD added and the corresponding response factor. In the case that blanks showed an artificial signal for the ISTD, it was taken adequately into account and the calculated amount was accordingly corrected.
Results and discussion
Approach
The original method according to Jezussek and Schieberle [29] based on an AA derivatization with 2-mercaptobenzoic acid and SIDA via LC–MS was optimized for a reliable quantitation of AA in vegetable chips. Its suitability was assessed by means of LoD, LoQ, and recovery rates. Vegetable chips samples from the worldwide market were analyzed to monitor their current AA amounts and those were compared to the current AA levels in potato chips, to which successful mitigation strategies have been applied for nearly 20 years.
Method development
To have a suitable AA quantitation method for vegetable chips at hand, the original approach based on the conversion of AA and the corresponding stable isotopically labeled ISTD [13C3]-AA into their respective stable thioethers (Fig. 1) was modified due to huge/coeluting matrix effects that hindered a reliable AA quantitation based on an S/N ratio of 10:1 in the analyzed samples. To identify differences in matrix loads during method optimization and to determine the recovery rate of AA, the extracts were analyzed by LC–MS both in the scan mode and the SIM mode, using a sweet potato chips sample (with and without the addition of AA and [13C3]-AA), a standard mixture of AA and [13C3]-AA, and a reagent blank.

Derivatization reaction of AA and [13C3]-AA (13C marked with black squares) with 2-mercaptobenzoic acid into a stable thioether according to Jezussek and Schieberle [29]
First, the extract obtained by the original method was additionally subjected to protein precipitation (Carrez clarification) commonly reported in literature [25] and dispersive solid phase extraction (dSPE) with charcoal as suggested by Granvogl and Schieberle [30] for the quantitation of AA in cocoa and coffee; however, both approaches did not reduce the matrix interferences in the present study. Next, a modified QuEChERS method was tested for the extraction of AA from the vegetable chips matrices. According to the method of Mastovska and Lehotay [37] for the analysis of AA in French fries, extraction and defatting was done with a mixture of acetonitrile/water/n-hexane (10/10/5, v/v/v), and phase separation was obtained by the addition of MgSO4 and NaCl. An additional clean-up step by dSPE both with (i) primary secondary amine (PSA; according to [37]) and (ii) graphite carbon black (an additional option within the original QuEChERS method) did only marginally reduce the matrix effects, the latter even led to worse recovery rates; thus, this purification step was omitted.
Therefore, SPE as a more powerful purification technique was used to clean-up the sample extract prior to the derivatization step. Several materials discussed in literature [18, 25,26,27,28, 38, 39] were considered, of which a hydrophilic-lipophilic balanced (HLB) reversed-phase sorbent consisting of an N-vinylpyrrolidone-divinylbenzene copolymer and a mixed-mode strong cation-exchange (MCX) reversed-phase polymer were tested in the present study. HLB cartridges should offer the retention of a broad range of polar and non-polar compounds and the MCX sorbent the retention of polar components and bases. Thereby, the MCX material was the most appropriate one concerning a minimum of matrix in the eluate and the highest recovery rate of AA. The final SPE consisted of a simple pass-through procedure of the defatted QuEChERS acetonitrile extract and a rinsing step to obtain a complete elution of AA and [13C3]-AA. It is noteworthy that the conversion into the stable thioethers was negatively affected by the presence of acetonitrile, why the solvent had completely to be removed prior to the derivatization step with 2-mercaptobenzoic acid.
During optimization of the subsequent LLE, dichloromethane was shown as the more suitable solvent compared to ethyl acetate used in the original method enhancing the recovery rate of the AA thioether and less matrix interferences were obtained. After the evaluation of shaking time and intensity and the number of repetitions, the final LLE was a threefold shaking with dichloromethane on a mechanical shaker for 10 min at RT.
Method sensitivity, precision, and recovery rate
The sensitivity of the procedure was assessed by LoQ in the reagent blank, which was determined according to the DIN 32645 calibration graph method [36], demanding a calibration that is close to the presumed LoD. Therefore, calibration curves were performed (n = 4) in the range of 15–150 µg of AA/L (analyzed as AA thioether), revealing good linearity with high correlation coefficients (R2 ≥ 0.9922) and good intraday and interday precisions, and leading to an LoQ of 33 µg of AA/L in the reagent blank. In addition, considering the real matrix effects, LoD (based on S/N ratio of 3:1) and LoQ (10:1) were estimated for all types of vegetable chips to 12 and 41 µg of AA/kg, which is below the EFSA requirement for an LoQ of 50 µg/kg outlined in 2015 for potato chips, coffee, and other products [13].
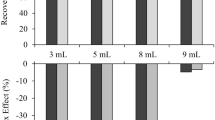
In a first trial, AA recovery experiments in vegetable chips were performed at levels of 100 and 500 µg/kg (n = 2) using a parsnip chips sample with a low native AA content of 100 µg/kg. In the next trial, recovery experiments were performed at levels of 1500 and 3000 µg/kg (n = 3) using a parsnip chips sample with a native AA content < LoQ. A reagent blank and a blank sample (without the addition of the ISTD) were additionally analyzed for both approaches and considered for calculation. Good recovery rates of 92% and 97% for the low AA levels with relative standard deviations (RSD) of 4% and 7% and of 100% and 101% for the high AA levels with RSD of 2% and 1% showed no considerable differences between the spiking levels. Thus, well repeatable results expressed by low RSD and high recovery rates showed the satisfying precision and accuracy of the entire method.
Acrylamide levels in vegetable chips from the worldwide market
The AA contents in 38 vegetable chips samples from different sources were determined. In total, 10 samples of sweet potatoes, 10 samples of parsnips, 11 samples of beetroots, and 7 samples of carrots were investigated. Figure 2 illustrates the mass traces obtained by LC–MS analysis representing the AA thioether (m/z 226) and the ISTD [13C3]-AA thioether (m/z 229), exemplarily shown for a calibration standard (A), a sweet potato chips sample containing 1210 µg of AA/kg (B), and a reagent blank (C). In addition, 12 samples of potato chips were analyzed for comparison. Results were well repeatable with RSD < 10% (n = 2 or 4 for each chips sample) (Table 1).

Mass traces obtained by LC–MS analysis representing the AA thioether (m/z 226) and the ISTD [13C3]-AA thioether (m/z 229), exemplarily shown for a calibration standard (corresponding to 5 mg of AA/L, equivalent to 833 µg of AA/kg sample) (A), a sweet potato chips sample containing 1210 µg of AA/kg (B), and a reagent blank (C)
The minimum content of 77.3 µg of AA/kg was detected in a beetroot sample and the maximum content of 3090 µg of AA/kg in a carrot sample. The sweet potato chips and the parsnip chips samples showed highest deviations within the same type of vegetable with factors of 28 and 27, respectively, between the lowest and the highest contents, and AA amounts ranging between 100 and 2780 µg of AA/kg and between 108 and 2870 µg of AA/kg, respectively (n = 10 for both types of vegetables). The variation within the beetroot chips samples was slightly lower with a factor of 20 and amounts between 77.3 and 1560 µg of AA/kg (n = 11), while the carrot chips samples showed a factor of only ~ 4 in a range between 700 and 3090 µg of AA/kg (n = 7) but the highest average content with 1390 µg of AA/kg among the vegetable chips.
Figure 3 visualizes that the relative distribution of AA in the different vegetable chips clearly depended on the type of vegetable and considerably differed within the four investigated types. Parsnip and sweet potato chips revealed rather low contents (< 750 µg of AA/kg) for most of the samples and showed only for a few samples very high contents (> 1500 µg of AA/kg), while carrot (except for 2 samples) and beetroot chips samples showed rather medium contents (600–1200 µg of AA/kg) for most of the samples. In total, 53% of the samples (20 of the 38 vegetable chips) exceeded the benchmark level of 750 µg of AA/kg stated by the EC for potato chips [14], which was slightly lower in comparison to the data obtained by Elmore et al. [22], who found higher contents than the set benchmark level for potato chips in 24 of 35 (69%) analyzed vegetable chips, bought in 2016. While 73% of the beetroot and 86% of the carrot chips samples exceeded the EU benchmark level, only 30% of the parsnip and of the sweet potato chips samples contained AA amounts > 750 µg/kg in the present study.

Percentage distribution of the acrylamide (AA) contents in 10 sweet potato chips samples (A), 10 parsnip chips samples (B), 11 beetroot chips samples (C), and 7 carrot chips samples (D)
The low amounts of free asparagine and reducing sugars in parsnips [22, 40,41,42,43] correlated well with the quite low contents of AA in these chips, and the high contents of reducing sugars in carrots [42] might be the reason for the quite high AA amounts in the carrot chips with a maximum amount of 3090 µg of AA/kg in one sample. However, the high contents of free asparagine and reducing sugars [22, 42] in sweet potatoes are not reflected in the medium average AA content in these chips (880 µg of AA/kg, n = 10). In detail, it was noticed that single-origin sweet potato chips showed rather low AA contents (average: 447 µg of AA/kg, n = 4), while sweet potato chips from chips mixtures contained high AA amounts (average: 1150 µg of AA/kg, n = 6). This might indicate successful mitigation strategies during the chips production of the single-origin chips. The relatively high AA levels in the beetroot chips samples did not correlate with the rather low amounts of free asparagine and reducing sugars in this vegetable [22, 42, 43]. Inappropriate processing parameters applied during the production of the mixed samples might be a reason. This assumption is supported by the fact that nearly all of the beetroot samples with elevated AA amounts (nine out of ten) were from mixed vegetable chips and by the already abovementioned finding of also rather high AA contents for sweet potato chips stemming from the same mixtures.
In potato chips, the average AA content was 449 µg/kg (matching well the data recently published by Powers et al. [17]), which was about half of the average amount of vegetable chips (963 µg/kg). Within the twelve potato chips samples investigated, the contents ranged between 117 and 832 µg of AA/kg and only two (17%) exceeded (one tightly, the other one clearly) the EU benchmark level. These data nicely corroborated that mitigation strategies for potato chips have clearly reduced their AA contents during the past two decades.
All in all, considering the average AA contents in vegetable chips, a trend for a slight reduction in the AA amounts compared to the dataset of the 2016 samples [22] was recognizable. However, 53% of the samples still exceeded the EU benchmark level for potato chips, why mitigation strategies should urgently be applied to follow the initial idea to be “healthy” chips. Furthermore, it is very important to distinguish between different vegetables based on the finding that the percentage of samples with AA amounts > 1000 µg/kg were clearly higher for beetroot and carrot chips compared to parsnip and sweet potato chips.
Conclusions
The derivatization of AA with 2-mercaptobenzoic acid into a stable thioether prior to analysis based on SIDA and LC–MS was shown as a reliable and sensitive method for AA quantitation in vegetable chips. A modified QuEChERS extraction including a defatting with n-hexane was followed by an SPE clean-up on strong cation-exchange material prior to derivatization. The application of [13C3]-AA as ISTD guaranteed reliable results as confirmed by recovery rates close to 100% at four spiking levels. With LoD and LoQ of 12 and 41 µg of AA/kg for the analyzed vegetable chips, a sufficient sensitivity was ensured allowing the determination of AA below the required LoQ of 50 µg of AA/kg, recommended by EFSA. The AA amounts of 38 vegetable chips samples from the worldwide market ranged between 77.3 and 3090 µg/kg. In general, carrots and beetroots showed elevated contents, while parsnips and sweet potatoes revealed rather low contents, with only few exceptions showing very high contents (> 1500 µg AA/kg). The average AA content in vegetable chips samples of 963 µg/kg clearly exceeded the benchmark level of the EC for potato chips and was twice as high compared to the average level of twelve potato chips samples (449 µg/kg). These data clearly demonstrated the successful application of mitigation strategies to potato chips during the past two decades on the one hand, but also the urgent need of such measures for this relatively new category of fried vegetable food on the other hand.
References
Tareke E, Rydberg P, Karlsson P, Eriksson S, Törnqvist M (2000) Acrylamide: a cooking carcinogen? Chem Res Toxicol 13:517–522. https://doi.org/10.1021/tx9901938
Tareke E, Rydberg P, Karlsson P, Eriksson S, Törnqvist M (2002) Analysis of acrylamide, a carcinogen formed in heated foodstuffs. J Agric Food Chem 50:4998–5006. https://doi.org/10.1021/jf020302f
Rosen J, Hellenas K-E (2002) Analysis of acrylamide in cooked foods by liquid chromatography tandem mass spectrometry. Analyst 127:880–882. https://doi.org/10.1039/b204938d
Mottram DS, Wedzicha BL, Dodson AT (2002) Acrylamide is formed in the Maillard reaction. Nature 419:448–449. https://doi.org/10.1038/419448a
Stadler RH, Blank I, Varga N, Robert F, Hau J, Guy PA, Robert M-C, Riediker S (2002) Acrylamide from Maillard reaction products. Nature 419:449–450. https://doi.org/10.1038/419449a
Zyzak DV, Sanders RA, Stojanovic M, Tallmadge DH, Eberhart BL, Ewald DK, Gruber DC, Morsch TR, Strothers MA, Rizzi GP, Villagran MD (2003) Acrylamide formation mechanism in heated foods. J Agric Food Chem 51:4782–4787. https://doi.org/10.1021/jf034180i
Granvogl M, Schieberle P (2006) Thermally generated 3-aminopropionamide as a transient intermediate in the formation of acrylamide. J Agric Food Chem 54:5933–5938. https://doi.org/10.1021/jf061150h
International Agency for Research on Cancer, World Health Organization (1994) IARC Monographs on the evaluation for carcinogenic risk of chemicals to humans: Some industrial chemicals, vol 60. https://publications.iarc.fr/78. Accessed 12 July 2021
Faria M, Ziv T, Gómez-Canela C, Ben-Lulu S, Prats E, Novoa-Luna KA, Admon A, Piña B, Tauler R, Gómez-Oliván LM, Raldúa D (2018) Acrylamide acute neurotoxicity in adult zebrafish. Sci Rep 8:e7918. https://doi.org/10.1038/s41598-018-26343-2
Belhadj Benziane A, Dilmi Bouras A, Mezaini A, Belhadri A, Benali M (2019) Effect of oral exposure to acrylamide on biochemical and hematologic parameters in Wistar rats. Drug Chem Toxicol 42:157–166. https://doi.org/10.1080/01480545.2018.1450882
Hogervorst JGF, de Bruijn-Geraets D, Schouten LJ, van Engeland M, de Kok TMCM, Goldbohm RA, van den Brandt PA, Weijenberg MP (2014) Dietary acrylamide intake and the risk of colorectal cancer with specific mutations in KRAS and APC. Carcinogenesis 35:1032–1038. https://doi.org/10.1093/carcin/bgu002
European Commission (2013) Commission Recommendation of 8 November 2013 on investigations into the levels of acrylamide in food. Off J Eur Union L 301:15–17
Scientific Committee EFSA, Benford D, Ceccatelli S, Cottrill B, DiNovi M, Dogliotti E, Edler L, Farmer P, Fürst P, Hoogenboom L, Knutsen HK, Lundebye A-K, Metzler M, Mutti A, Nebbia CS, O’Keeffe M, Petersen A, Rietjens I, Schrenk D, Silano V, van Loveren H, Vleminckx C, Wester P (2015) Scientific Opinion on acrylamide in food. EFSA J 13:4104. https://doi.org/10.2903/j.efsa.2015.4104
European Commission (2017) Commission Regulation (EU) 2017/2158 of 20 November 2017 establishing mitigation measures and benchmark levels for the reduction of the presence of acrylamide in food. Off J Eur Union L 304:24–44
Olsson K, Svensson R, Roslund C-A (2004) Tuber components affecting acrylamide formation and colour in fried potato: variation by variety, year, storage temperature and storage time. J Sci Food Agric 84:447–458. https://doi.org/10.1002/jsfa.1681
de Wilde T, de Meulenaer B, Mestdagh F, Govaert Y, Vandeburie S, Ooghe W, Fraselle S, Demeulemeester K, van Peteghem C, Calus A, Degroodt J-M, Verhé R (2006) Influence of fertilization on acrylamide formation during frying of potatoes harvested in 2003. J Agric Food Chem 54:404–408. https://doi.org/10.1021/jf0521810
Powers SJ, Mottram DS, Curtis A, Halford NG (2017) Acrylamide levels in potato crisps in Europe from 2002 to 2016. Food Addit Contam Part A 34:2085–2100. https://doi.org/10.1080/19440049.2017.1379101
U.S. Food and Drug Administration. Survey data on acrylamide in food: individual food products (2019) https://www.fda.gov/food/chemicals/survey-data-acrylamide-food/. Accessed 12 July 2021
Hamlet CG, Asuncion L, Liang L (2014) Survey of acrylamide and furan in UK retail products: summary report for samples purchased between November 2011 and Dec 2013. Food Standards Agency C030, pp 1–74
Hamlet CG, Liang L, Baxter B, Apostilova D, Rozeela A (2019) Survey of acrylamide and furans in UK retail products: summaries and trends for samples purchased between January 2014 and November 2018. Food Standards Agency C041, pp 1–51
Changing Markets (2017) High levels of carcinogenic chemical, acrylamide, found in potato crisps from major UK brands and retailers. Changing Markets, London. http://changingmarkets.org/wp-content/uploads/2017/03/ACRYLAMIDE-UK-potato-snacks-press-release-results-FINAL.pdf. Assessed 12 July 2021
Elmore JS, Xu F, Maveddat A, Kapetanou R, Qi H, Oruna-Concha M-J (2019) Acrylamide content of vegetable chips. In: Granvogl M, MacMahon S (eds) Food-borne toxicants: formation, analysis, and toxicology. American Chemical Society, Washington, pp 15–26
Breitling-Utzmann CM, Hankele S (2019) Formation of acrylamide in vegetable crisps – Influence of processing conditions and reducing sugars. Dtsch Lebensm Rundsch 115:265–270
European Commission (2019) Commission Recommendation (EU) 2019/1888 of 7 November 2019 on the monitoring of the presence of acrylamide in certain foods. Off J Eur Union L 290:31–33
Wenzl T, de la Calle MB, Anklam E (2003) Analytical methods for the determination of acrylamide in food products: a review. Food Addit Contam 20:885–902. https://doi.org/10.1080/02652030310001605051
Keramat J, LeBail A, Prost C, Soltanizadeh N (2011) Acrylamide in foods: chemistry and analysis. A review. Food Bioprocess Technol 4:340–363. https://doi.org/10.1007/s11947-010-0470-x
Elbashir AA, Omar MMA, Ibrahim WAW, Schmitz OJ, Aboul-Enein HY (2014) Acrylamide analysis in food by liquid chromatographic and gas chromatographic methods. Crit Rev Anal Chem 44:107–141. https://doi.org/10.1080/10408347.2013.829388
Arvanitoyannis IS, Dionisopoulou N (2014) Acrylamide: formation, occurrence in food products, detection methods, and legislation. Crit Rev Food Sci Nutr 54:708–733. https://doi.org/10.1080/10408398.2011.606378
Jezussek M, Schieberle P (2003) A new LC/MS-method for the quantitation of acrylamide based on a stable isotope dilution assay and derivatization with 2-mercaptobenzoic acid. Comparison with two GC/MS methods. J Agric Food Chem 51:7866–7871. https://doi.org/10.1021/jf0349228
Granvogl M, Schieberle P (2007) Quantification of 3-aminopropionamide in cocoa, coffee and cereal products. Eur Food Res Technol 225:857–863. https://doi.org/10.1007/s00217-006-0492-9
Tölgyesi Á, Sharma VK (2020) Determination of acrylamide in gingerbread and other food samples by HILIC-MS/MS: a dilute-and-shoot method. J Chromatogr B 1136:121933. https://doi.org/10.1016/j.jchromb.2019.121933
Yoshioka T, Izumi Y, Nagatomi Y, Miyamoto Y, Suzuki K, Bamba T (2019) A highly sensitive determination method for acrylamide in beverages, grains, and confectioneries by supercritical fluid chromatography tandem mass spectrometry. Food Chem 294:486–492. https://doi.org/10.1016/j.foodchem.2019.05.033
Gökmen V, Morales FJ, Ataç B, Serpen A, Arribas-Lorenzo G (2009) Multiple-stage extraction strategy for the determination of acrylamide in foods. J Food Compos Anal 22:142–147. https://doi.org/10.1016/j.jfca.2008.09.007
Jozinović A, Šarkanj B, Ačkar D, Balentić JP, Šubarić D, Cvetković T, Ranilović J, Guberac S, Babić J (2019) Simultaneous determination of acrylamide and hydroxymethylfurfural in extruded products by LC-MS/MS method. Molecules 24:1971. https://doi.org/10.3390/molecules24101971
Stiftung Warentest (2017) Gemüsechips—Kritische Stoffe verderben den Knabberspaß. https://www.test.de/Test-Gemuesechips-5220648-5220653. Accessed 12 July 2021
Deutsches Institut für Normung (2008) DIN 32645 Chemical analysis - Decision limit, detection limit and determination limit under repeatability conditions - Terms, methods, evaluation. Beuth Verlag, Berlin
Mastovska K, Lehotay SJ (2006) Rapid sample preparation method for LC-MS/MS or GC-MS analysis of acrylamide in various food matrices. J Agric Food Chem 54:7001–7008. https://doi.org/10.1021/jf061330r
Young MS, Jenkins KM, Mallet CR (2004) Solid-phase extraction and cleanup procedures for determination of acrylamide in fried potato products by liquid chromatography/mass spectrometry. J AOAC Int 87:961–964
Liu J, Zhao G, Yuan Y, Chen F, Hu X (2008) Quantitative analysis of acrylamide in tea by liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Food Chem 108:760–767. https://doi.org/10.1016/j.foodchem.2007.11.015
Takahata Y, Noda T, Nagata T (1995) Varietal differences in crude protein, and free and hydrolyzed amino acid compositions of sweetpotato roots. J Jpn Soc Food Sci Technol 42:362–368. https://doi.org/10.3136/nskkk.42.362
Nandula VK, Foster JG, Foy CL (2000) Impact of Egyptian broomrape (Orobanche aegyptiaca (Pers.) parasitism on amino acid composition of carrot (Daucus carota L.). J Agric Food Chem 48:3930–3934. https://doi.org/10.1021/jf991145w
Souci SW, Fachmann W, Kraut H (2000) Food composition and nutrition tables, 6th edn. Medpharm, Stuttgart
Kugler F, Graneis S, Schreiter PP-Y, Stintzing FC, Carle R (2006) Determination of free amino compounds in betalainic fruits and vegetables by gas chromatography with flame ionization and mass spectrometric detection. J Agric Food Chem 54:4311–4318. https://doi.org/10.1021/jf060245g
Acknowledgements
The authors would like to express many thanks to Erasmus student Miss Sofia Ourda and master’s student Miss Annelie Kehrer for support in the laboratory.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Compliance with ethics requirements
The authors Claudia Oellig, Eva Gottstein, and Michael Granvogl hereby confirm that this manuscript is performed according and follows the COPE guidelines and has not already been published nor is it under consideration for publication elsewhere. This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oellig, C., Gottstein, E. & Granvogl, M. Analysis of acrylamide in vegetable chips after derivatization with 2-mercaptobenzoic acid by liquid chromatography–mass spectrometry. Eur Food Res Technol 248, 937–946 (2022). https://doi.org/10.1007/s00217-021-03898-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-021-03898-5