
Abstract
Nucleic acid detection technology based on polymerase chain reaction (PCR) and antibody detection based on immunochromatography still have many problems such as false negatives for the diagnosis of coronavirus disease 2019 (COVID-19). Therefore, it is of great importance to develop new techniques to improve the diagnostic accuracy of COVID-19. We herein developed an ultrasensitive, rapid, and duplex digital enzyme-linked immunosorbent assay (dELISA) for simultaneous detection of spike (S-RBD) and nucleocapsid (N) proteins of SARS-CoV-2 based on a single molecule array. This assay effectively combines magnetic bead encoding technology and the ultrasensitive detection capability of a single molecule array. The detection strategies of S-RBD protein and N-protein exhibited wide response ranges of 0.34–1065 pg/mL and 0.183–338 pg/mL with detection limits of 20.6 fg/mL and 69.8 fg/mL, respectively. It is a highly specific method for the simultaneous detection of S-RBD protein and N-protein and has minimal interference from other blood proteins. Moreover, the spike assay showed a satisfactory and reproducible recovery rate for the detection of S-RBD protein and N-protein in serum samples. Overall, this work provides a highly sensitive method for the simultaneous detection of S-RBD protein and N-protein, which shows ultrasensitivity and high signal-to-noise ratio and contributes to improve the diagnosis accuracy of COVID-19.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The outbreak of the novel coronavirus disease 2019 (COVID-19) occurred in late 2019, and now has spread worldwide resulting in a global pandemic [1]. COVID-19 is caused by a novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which has infected more than 164 million people and resulted in more than 3.4 million deaths. The high morbidity and mortality of COVID-19 have far exceeded those of seasonal influenza and other diseases [2,3,4]. At present, SARS-CoV-2 is still spreading globally, causing long-term effects on human health and normal activities [5]. Since no specific medicine or treatment for COVID-19 is available, the accurate diagnosis and a series of prevention and control measures have become the most effective means to prevent its spread.
Current diagnostic approaches mainly include two categories: nucleic acid testing based on RT-PCR technology and antibody testing based on immunochromatography [6,7,8,9,10,11]. The RNA test of SARS-CoV-2 pioneered by the Centers for Disease Control (CDC) has been deemed the “gold standard” for clinical diagnosis. However, drawbacks such as long hours to perform, need for specialized reagents, equipment, and trained operators restrict its application on a large scale [12, 13]. Except for RT-PCR, two isothermal techniques have been developed by researchers for the rapid and sensitive detection of viral RNA including loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA) [14,15,16,17,18]. However, previous studies showed that the positive rate of viral RNA testing is only 30–60%, which suggests a high false-negative rate of nucleic acid detection for COVID-19 [12, 19,20,21]. Several limitations and issues require further research such as different respiratory samples, inappropriate sample collection, transfer, and processing [22, 23]. On the other hand, degradation of purified RNA, the presence of RT-PCR inhibitors, or genomic mutations may cause false-negative results [22, 24, 25]. In addition, the immunoassay has also been used to detect the antibodies that are created by our body in response to SARS-CoV-2 infection [12, 22, 26]. Detection of microbe-specific IgM and IgG antibodies in circulating blood serves as a traditional method to identify whether a person has been infected with the pathogen [27]. In COVID-19, IgM and IgG antibodies can arise nearly simultaneously in serum within 2 to 3 weeks after the onset of illness [9, 28, 29]. Several detection methods for detecting IgG and IgM have been developed as rapid diagnosis of COVID-19 such as ELISA and magnetic chemiluminescence immunoassay [30,31,32,33,34]. Liu et al. used two ELISA kits based on SARS-CoV-2 spike and nucleocapsid proteins to detect IgM and IgG antibodies and evaluated their diagnostic feasibility [35]. Chen et al. reported a rapid and sensitive lateral flow immunoassay that used lanthanide-doped polystyrene nanoparticles to detect anti-SARV-CoV-2 IgG in human serum [36]. Although antibody testing has also been used in the auxiliary diagnosis of COVID-19, some tests may cross-react with other coronaviruses, such as those that cause the common cold [37]. Therefore, the development of new techniques with an improved diagnostic accuracy of COVID-19 is in high demand. The spike (S) and nucleocapsid (N) proteins of SARS-CoV-2 are two promising antigen biomarkers for the diagnosis of COVID-19 in human blood as they play key roles in the receptor recognition, virus replication, and immune response [38,39,40]. Many experts believe that the detection of viral protein antigens could be helpful for the diagnosis of COVID-19 in accordance with the SARS-CoV protein antigen detection experience [41,42,43]. Unlike exponential amplification of the nucleic acid detection, proteins cannot be directly amplified, thus the detection of minute amount of proteins demands ultrasensitive detection techniques [12].
Single molecule array (Simoa) is a digital ELISA [44]. Simoa was developed by David Walt’s group for the detection of proteins with extremely high analytical sensitivity, which can be 1000 times higher than that of traditional ELISAs. In digital ELISA, the fluorescence produced by the enzyme−substrate reaction is confined into femtoliter-sized microwells. Since each microwell can only fit one bead, the presence of a single-protein molecule can be detected via fluorescence read-out [45]. Herein, in this work, we have proposed a digital ELISA method to simultaneously and ultrasensitively detect S-RBD protein and N-protein via Simoa and the magnetic bead encoding technology. This work identifies the most reliable antibody pairs to detect two proteins by the selection process and performs the optimization of the Simoa reagents for the highest sensitivity and dynamic range using recombinant proteins. Furthermore, the proposed assay has high potential for the diagnosis of COVID-19 and offers opportunities to monitor the patients by testing S-RBD protein and N-protein in the blood.
Materials and methods
Materials and instruments
Recombinant S-RBD protein (40592-V08B) with a molecular weight (MW) of 26.54 kDa and N-protein (40588-V08B) with a MW of 47.08 kDa, monoclonal antibodies (mAbs) (S-RBD protein mAbs, SA1(MM43), SA2(D006), SA3(R0013); N-protein mAbs, NA1(R001), NA2(R004), NA3(MM05), NA4(R040), NA5(R019)), MERS-CoV spike protein and nucleoprotein protein were provided by SinoBiological (Bei**g, China). Two S-RBD protein antibodies (SA4(NBP2-90980), SA5(NBP2-90979)), carcinoembryonic antigen (CEA), and angiotensin-converting enzyme 2 (ACE2) were bought from Bio-Techne China (Shanghai, China). Cyfra21-1 antigen was purchased from Fitzgerald (USA). Alpha-fetoprotein was provided from Linc-Bio (China). Procline 300, phosphate buffer saline (PBS, 10X), Tween20, Ethylenediaminetetraacetic acid disodium salt (EDTA), and 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide hydrochloride (EDC) were purchased from Sigma-Aldrich (MO, USA). NHS-PEG4-Biotin was bought from ThermoFisher Scientific (MA, USA). Newborn calf serum was purchased from Sijiqing (Hangzhou, China). Sample diluent was provided by Bright Spot Biotechnology (Zhengzhou, China). Carboxylated paramagnetic beads (MB-COOH), 488-labeled and 700-labeled beads (d = 2.7 μm), disc kit, bead conjugation buffer, bead wash buffer, bead diluent, biotinylation buffer, detector diluent, calibrator diluent, wash buffer1, wash buffer2, streptavidin-β-galactosidase (SBG), SBG diluent, and resorufin-β-D-galactopyranoside (RGP) were obtained from Quanterix (Lexington, USA).
The 96-well plate and the constant temperature incubator shakers were purchased from IKA (Germany) and Eppendorf (Germany) for antibody pair selection and bead conjugation, respectively. NanoDrop One Spectrophotometer (ThermoFisher Scientific, USA) was used to measure the concentration of the antibodies and evaluated bead coupling efficiency. Microplate reader (Tecan, Switzerland) measured fluorescent intensity of 96-well plate when antibody pairs were selected by ELISA. Simoa HD-X analyzer (Quanterix, USA) was used to conduct the detection assay.
Bioconjugation of MB-COOH with S and N protein mAbs
MB-mAbs conjugates were prepared following the protocol in a previous report [46]. In the duplex assay, 488-labeled MB and 700-labeled MB were chosen to conjugate S-RBD protein mAbs and N-protein mAbs, respectively. In brief, 285 μL of the MB-COOH (1.5 × 109 beads/mL) and 15 μL of the EDC (10 mg/mL) were added to a 1.7-mL microcentrifuge tube, and then the mixture was stirred for 30 min at 4 °C in a shaking incubator. Next, 80 μL of the mAbs (1.0 mg/mL) in bead conjugation buffer was added into the reaction. The mixture was allowed to react for another 4 h at 4 °C in a shaking incubator. Afterward, MB-mAbs were blocked in bead block buffer for 30 min at room temperature. The conjugated compounds were washed by bead wash buffer and bead diluent. The final MB-mAbs were transferred into a new microcentrifugation tube and stored in bead diluent at 4 °C for further use.
Biotinylation of detection antibody
To enable the detection antibody to bind specifically to SBG tightly, the detection antibody needs to be labeled with biotin. In brief, 50 μg of detection antibody was reconstituted with PBS. Then, 1.5 μL of NHS-PEG4-Biotin (8.9 mM) was added to the detection antibody solution and reacted at room temperature for 30 min. The biotin-labeled detection antibody was prepared by the coupling reaction between the N-hydroxysuccinimide ester (NHS) group of NHS-PEG4-Biotin and the amino group of antibodies. The concentration of the purified detection antibodies was determined by a NanoDrop One spectrophotometer. The final antibody was aliquoted and stored at −20 °C.
MB-based ELISA for the selection of antibody pairs
To select the optimal antibody pairs for the ultrasensitive detection of S and N proteins, a traditional ELISA based on magnetic beads was performed. The 100 μL of MB-capture mAbs (500,000 beads) was added into a 96-well plate. After MB were magnetically separated and washed, 100 μL of the different concentrations (0, 1, 10, 100 ng/mL) of recombinant S-RBD protein or N-protein was added into the corresponding well. The mixture was incubated at room temperature under gentle shaking for 1 h. Then the mixture was placed on a magnetic rack, separated, and washed three times. Afterward, the detection antibodies (0.3 μg/mL) and SBG (150 pM) were added to the reaction system in order for 1 h and 0.5 h incubation, followed by separation and washing procedures. The isolated immunocomplex was washed three times with wash buffer1. The isolated immunocomplex was re-dispersed in 100 μL of 100 μM RGP solution and incubated at room temperature for 15 min. The fluorescence intensity of the well was measured by a microwell reader with an excitation and emission wavelength of 555 nm and 595 nm.
Simoa assay for the detection of S-RBD protein and N-protein
Singleplex and duplex Simoa assays were performed for the ultrasensitive detection of S-RBD protein and N-protein on Simoa HD-X Analyzer. The assay operation procedures of the two modes were similar. The beads with capture mAbs were diluted in calibrator diluent and about 500,000 per test. The detection antibodies of S-RBD protein and N-protein were diluted to 0.5 μg/mL in detector diluent, respectively. SBG was diluted to 400 pM in SBG diluent. Recombinant S-RBD protein and N-protein were serially diluted and served as standards. The singleplex and duplex protocols of “2-step assay neat with 50 μL RGP” were set up, respectively. Differently, the bead type of the singleplex assay was set to 488L0. The bead types of S-RBD protein and N-protein in the duplex assay were set to 488L1 and 700L1, respectively. (L0 and L1 represent a specification of magnetic beads, which match the parameters on the Analyzer.) In the first step of this assay, the capture mAbs-coated magnetic beads, the biotinylated detection antibody, and the calibrators or the samples were mixed and reacted for 35 min in cuvette. After the washing process, the SBG solution was added to the cuvette and incubated for 5 min to form an immunocomplex. Then, the RGP solution was mixed with the immunocomplex in cuvette and loaded into the disc microarray. Finally, the AEB (average enzymes per bead) value was recorded as signal.
Specificity investigation
To assess the response of other blood proteins, different interfering blood proteins, including CEA (R&D), Cyfra21-1 (Fitzgerald), ACE2 (R&D), and AFP (Linc-Bio), were tested by two proposed singleplex Simoa assays. The detection process was the same as the previous part.
The spike and recovery assay
In order to verify the detection ability of the proposed assay in serum and saliva samples, the spike and recovery assay was performed. Firstly, the healthy human blood samples were collected in plain tube. Serum sample was obtained by centrifugation at 4000×g for 5 min. Serum and saliva sample was diluted 4-fold and 8-fold using sample diluent and was spiked with different concentrations of S-RBD protein and N-protein. The detection procedure was the same as the previous part.
Results and discussion
Singleplex and duplex Simoa assays for the detection of S-RBD protein and N-protein
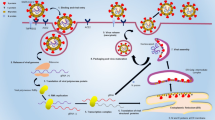
This work uses double-antibody sandwich structure and single molecule array technology to develop a duplex assay for the ultrasensitive detection of the SARS-CoV-2 S-RBD protein and N-protein. First of all, the capture mAbs-coated magnetic beads were prepared by EDC coupling. After coupling, the capture antibody and the carboxyl group of the MBs form an amide bond. Thus, the capture antibodies of S-RBD protein and N-protein can be strongly bound to the surface of the MBs by the covalent bonds. Then, the detection antibodies, which contain an available recognition site different from the capture antibody, can bind to other parts of the S-RBD protein and N-protein to form a sandwich-type structure. To achieve the binding of detection antibody and SBG, the detection antibodies are labeled using the biotinylated reagent. Due to biotin and streptavidin having a strong binding affinity, the MBs and SBG form a stable immunocomplex. RGP is the fluorescent substrate of SBG and has good water solubility. When SBG is present, the ether bond between β-galactose and resorufin can be broken, so that resorufin produces a strong fluorescence signal. The above is the conventional process of the classic enzyme-linked immunosorbent assay. However, the change of fluorescence signal which can be detected by microplate reader normally requires very high concentrations of enzymes in 100–300 μL of reaction volume. This mode limits the detection sensitivity because the weak signal intensity is difficult to detect at low concentrations of the target. When the enzymatic reaction is transferred to a microwell with about 46 fL, the signal generated by the catalytic substrate of a single enzyme molecule can be easily detected (Fig. 1a). In the Simoa assay, capture antibody-coated beads are added far more than the low concentrations of target protein molecules. On the basis of Poisson statistics, either one or zero target protein molecules will bind to each bead [45]. That is, one bead with a protein will capture an enzyme molecule. Since the diameter of the microwells in the array is about 4.25 μm, the microwell can accommodate only one magnetic bead with 2.7 μm diameter. After the magnetic bead immunocomplex was loaded into the microwell, the perfluorinated oil seals the microwell while removing the magnetic beads on the surface of the array. Then, a fluorescence microscope performs dark field imaging on the microwell array chip to determine the position and number of magnetic beads. The microwells containing the enzyme-labeled protein were fluorescently imaged and counted. According to this principle, magnetic bead ELISA combined with microwell imaging technology could greatly improve the detection sensitivity of the target. In addition, the high sensitivity of this method for the S-RBD protein and N-protein determination is also attributed to the Poisson distribution, where either one or zero target protein molecules will bind to each bead. According to the principle of Poisson distribution, when the number of proteins is far less than the total number of magnetic beads, the number of magnetic beads with more than one protein can be ignored. Within a certain concentration range, the signal value will increase with the concentration of S-RBD protein and N-protein. Finally, the quantitative detection of S-RBD protein and N-protein can be achieved this way.

Schematic illustration of the S-RBD protein and N-protein Simoa assay. a Schematic illustration of singleplex Simoa assay for the ultrasensitive detection of the S-RBD protein or N-protein. b Schematic illustration of duplex Simoa assay for the simultaneous ultrasensitive detection of the S-RBD protein and N-protein
To achieve the purpose of the simultaneous detection, two kinds of capture antibodies are coupled to the surface of magnetic beads with different fluorescent dyes in duplex Simoa assay (Fig. 1b). Other procedures are similar to the singleplex assay. The difference is that the magnetic beads of S-RBD protein (488 dye-labeled) and N-protein (700 dye-labeled) are respectively imaged and positioned under the corresponding excitation/emission filter. Finally, RGP fluorescence-based microwell array imaging can distinguish the respective targets via the previous beads’ positioning. Therefore, the simultaneous and ultrasensitive detection of S-RBD protein and N-protein can be achieved based on the above procedure.
Selection of the antibody pairs
In order to achieve the excellent detection performance, this assay paired the five antibodies with each other and screened the best combination for further test. The screening was performed by using the traditional MB-ELISA, and the detailed process was described in the experiment section. According to the design, a total of 20 antibody pair combinations for each protein were used to determine the different concentrations of S-RBD protein and N-protein. The best combinations were selected by comparing their fluorescence intensity at 0, 1, 10, and 100 ng/mL of S-RBD protein and N-protein. As shown in Supplementary information (ESM) Figs. S1 and S2, the SA2-SA1 and NA3-NA1 antibody pair combination shows the highest signal-to-noise ratio for the detection of S-RBD protein and N-protein, respectively. The results also proved that the selected antibodies could recognize different sites of the target proteins and exhibit high affinity to S-RBD protein and N-protein respectively. The selected antibody pair combinations were tested on Simoa HD-X Analyzer for further study.
Feasibility of the strategy
To verify the feasibility of simultaneous detection of S-RBD protein and N-protein, AEB values of S-RBD protein and N-protein at different concentrations on the chip and the position images of the magnetic beads were obtained (Fig. 2) [47, 48]. The images are processed by ImageJ software in this assay. As shown in Fig. 2a, the positions of the magnetic beads corresponding to the S-RBD protein and N-protein are located through the labeled fluorescent molecules and filters. Since each microwell can only fit one magnetic bead, the two magnetic beads do not overlap in Fig. 2a. In addition, the emission wavelength of the RGP substrate was observed with no interference caused by the two dyes. When performing concentration counting and quantification, two target proteins can be detected at the same time. As the concentration of S-RBD protein and N-protein increases, the number of bright dots increases significantly. At the same time, through statistical bright dots and simulation calculations, the corresponding AEB value at each concentration was obtained, as shown in Fig. 2b. The results indicate that the developed method was feasible for simultaneous detection of S-RBD protein and N-protein.

a S-RBD protein and N-protein correspond to local images of 488 dye- and 700 dye-labeled magnetic beads, respectively. b AEB values corresponding to S-RBD protein and N-protein at different concentrations (for replicates, n = 3)
Quantitative detection based on singleplex assay
In order to confirm that the developed method can achieve the ultrasensitive quantitative detection of S-RBD protein and N-protein, we first developed a singleplex Simoa assay to investigate the signal changes with more S-RBD protein and N-protein concentration gradients. Figure 3 showed the calibration curve when S-RBD protein and N-protein are detected separately based on a singleplex Simoa assay. There is a certain functional relationship between the concentration of S-RBD protein or N-protein and the AEB value. Through function fitting, it is found that the two conform to a four-parameter function equation [47]:

Calibration plots of a S-RBD protein and b N-protein based on the singleplex Simoa assay
In the equation, A, B, C, and D represent the fitting constant (Table 1), Y represents the AEB value, and x represents the concentration of S-RBD protein and N-protein. In addition, according to the principle of blank+2.5 times the standard deviation, the lower detection limits (LODs) of S-RBD protein and N-protein in the developed singleplex assay reached 34.2 fg/mL and 76.4 fg/mL, and their linear ranges were 0.217–269 pg/mL and 1.15–1438 pg/mL, respectively. This result laid the foundation for the development of the duplex Simoa assay.
Selectivity investigation and duplex Simoa assay
The selectivity of developed assay is of great significance for further use in complex media. In order to avoid false positive signals that may be caused by other diseases, we investigated several serum high-level proteins (ACE2, CEA, Cyfra21-1, AFP) and the MERS-related proteins that coexist in biosystems and may act as potential interferents. As shown in Fig. 4, the AEB value corresponding to 50 pg/mL S-RBD protein and 500 pg/mL N-protein is higher than that of other serum protein markers ACE2, CEA, Cyfra21-1, and AFP (5 ng/mL). This result indicated that two singleplex assays had good selectivity for the S-RBD protein and N-protein detection. In order to achieve the simultaneous detection of S-RBD protein and N-protein, the mutual interference between the two proteins is also investigated. The results confirmed that there were no obvious interference and cross-reaction between the two proteins in the detection of each other.

The cross-reaction study in the detection of a S-RBD protein and b N-protein based on a singleplex Simoa assay. S, S-RBD protein; N, N-protein; ACE2, angiotensin converting enzyme 2; CEA, carcinoembryonic antigen; Cyfra21-1, the fragment of cytokeratin 19; AFP, alpha fetoprotein; MERS-N, MERS nucleocapsid protein; MERS-S, MERS spike protein
On the basis of the above results, we developed a duplex Simoa assay for the simultaneous and ultrasensitive detection of S-RBD protein and N-protein. Figure 5 shows the calibration curve of the simultaneous detection of S-RBD protein and N-protein. The LODs of S-RBD protein and N-protein were estimated to be 20.6 fg/mL and 69.8 fg/mL with a wide response range of 0.34–1065 pg/mL and 0.183–338 pg/mL, respectively. The results demonstrate that there is no significant difference between the duplex and the singleplex assays in sensitivity and detection range, but it meets the requirements of blood SARS-CoV-2 antigen detection [39, 43]. Therefore, the duplex Simoa assay is developed for the simultaneous detection of S-RBD protein and N-protein, which facilitate accurate diagnosis of COVID-19. Compared to the singleplex assay, the duplex assay exhibits better application potential and value.

Calibration plots of S-RBD protein (blue line) and N-protein (red line) based on the duplex Simoa assay
Duplex assay validation
To evaluate the detection performance of the S-RBD protein and N-protein duplex Simoa assay in complex serum samples, a certain concentration of proteins and serum samples were mixed in a ratio of 1:1 and serially diluted. As shown in Fig. 6, a good linear relationship between the serum dilution factor and the S-RBD protein and N-protein concentrations was observed. This result indicates that the duplex Simoa can effectively measure the concentration of S-RBD protein and N-protein in serum samples.

Linear relationship between different serum dilution factors and detected S-RBD protein (blue line) and N-protein (red line) concentrations based on duplex Simoa assay
In addition, the recovery experiments were carried out to further verify the reliability of the method for simultaneous detection of S-RBD protein and N-protein in serum. Different concentrations of S-RBD protein and N-protein standards were added to the 4-fold diluted serum. As shown in Table 2, the 4-fold diluted serum was added with 0 pg/mL, 5 pg/mL, 20 pg/mL, and 80 pg/mL S-RBD protein and 0 pg/mL, 8 pg/mL, 80 pg/mL, and 800 pg/mL N-protein standards and the corresponding S-RBD protein and N-protein concentrations calculated according to the standard curve. The results showed that the recovery rates of S-RBD protein were 90.4%, 82.8%, and 88.3%, and the recovery rates of N-protein were 83.1%, 77.6%, and 98.7%, respectively. Furthermore, we also carried out spike recovery assays in saliva samples and achieved good results (ESM Table S1). The good recovery rate indicates that the developed duplex Simoa assay has high reliability, and it is promising to be used for the further detection of S-RBD protein and N-protein in the serum of COVID-19 patients. Although this method exhibited sufficient sensitivity, practical experiments with COVID-19 samples were not carried out in this study due to the shortage of COVID-19 samples in China.
Conclusion
In summary, the developed duplex single molecule assay demonstrates to be efficient for the simultaneous and ultrasensitive detection of S-RBD protein and N-protein from SARS-CoV-2. Furthermore, this assay shows the advantages of rapidness, high sensitivity, and excellent specificity, and the limits of detection for simultaneous detection of S-RBD protein and N-protein were estimated to be 20.6 fg/mL and 69.8 fg/mL with wide detection ranges of 0.34–1065 pg/mL and 0.183–338 pg/mL, respectively. This assay could not only successfully monitor S-RBD protein and N-protein in aqueous solution but also exhibit reliable responses toward the spiked proteins in human serum and saliva samples. This duplex assay provides a promising tool for detecting S-RBD protein and N-protein and has great potential for supplementary diagnosis of COVID-19. We hope that the global COVID-19 epidemic will be brought under control as soon as possible.
References
Jeyanathan M, Afkhami S, Smaill F, Miller MS, Lichty BD, **ng Z. Immunological considerations for COVID-19 vaccine strategies. Nat Rev Immunol. 2020;10:615–32. https://doi.org/10.1038/s41577-020-00434-6.
Sharma A, Tiwari S, Deb MK, Marty JL. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int J Antimicrob Agents. 2020;2:106054. https://doi.org/10.1016/j.ijantimicag.2020.106054.
Feng W, Zong W, Wang F, Ju S. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): a review. Mol Cancer. 2020;1:100. https://doi.org/10.1186/s12943-020-01218-1.
Bakouny Z, Hawley JE, Choueiri TK, Peters S, Rini BI, Warner JL, et al. COVID-19 and cancer: current challenges and perspectives. Cancer Cell. 2020. https://doi.org/10.1016/j.ccell.2020.09.018.
Zheng J. SARS-CoV-2: an emerging coronavirus that causes a global threat. Int J Biol Sci. 2020;10:1678–85. https://doi.org/10.7150/ijbs.45053.
Tang Y-W, Schmitz JE, Persing DH, Stratton CW. Laboratory diagnosis of COVID-19: current issues and challenges. J Clin Microbiol. 2020;6:e00512–20. https://doi.org/10.1128/JCM.00512-20.
Pokhrel P, Hu C, Mao H. Detecting the coronavirus (COVID-19). ACS Sens. 2020;8:2283–96. https://doi.org/10.1021/acssensors.0c01153.
Mathuria JP, Yadav R, Rajkumar. Laboratory diagnosis of SARS-CoV-2 - a review of current methods. J Infect Public Health. 2020;7:901–5. https://doi.org/10.1016/j.jiph.2020.06.005.
Long Q-X, Liu B-Z, Deng H-J, Wu G-C, Deng K, Chen Y-K, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. 2020;6:845–8. https://doi.org/10.1038/s41591-020-0897-1.
Li Y, Yao L, Li J, Chen L, Song Y, Cai Z, et al. Stability issues of RT-PCR testing of SARS-CoV-2 for hospitalized patients clinically diagnosed with COVID-19. J Med Virol. 2020;7:903–8. https://doi.org/10.1002/jmv.25786.
La Marca A, Capuzzo M, Paglia T, Roli L, Trenti T, Nelson SM. Testing for SARS-CoV-2 (COVID-19): a systematic review and clinical guide to molecular and serological in-vitro diagnostic assays. Reprod BioMed Online. 2020;3:483–99. https://doi.org/10.1016/j.rbmo.2020.06.001.
Feng W, Newbigging AM, Le C, Pang B, Peng H, Cao Y, et al. Molecular diagnosis of COVID-19: challenges and research needs. Anal Chem. 2020;15:10196–209. https://doi.org/10.1021/acs.analchem.0c02060.
Esbin MN, Whitney ON, Chong S, Maurer A, Darzacq X, Tjian R. Overcoming the bottleneck to widespread testing: a rapid review of nucleic acid testing approaches for COVID-19 detection. RNA (New York, NY). 2020;7:771–83. https://doi.org/10.1261/rna.076232.120.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. New Engl J Med. 2020;8:727–33. https://doi.org/10.1056/NEJMoa2001017.
Yu L, Wu S, Hao X, Dong X, Mao L, Pelechano V, et al. Rapid detection of COVID-19 coronavirus using a reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) diagnostic platform. Clin Chem. 2020;7:975–7. https://doi.org/10.1093/clinchem/hvaa102.
Baek YH, Um J, Antigua KJC, Park J-H, Kim Y, Oh S, et al. Development of a reverse transcription-loop-mediated isothermal amplification as a rapid early-detection method for novel SARS-CoV-2. Emerg Microb Infect. 2020;1:998–1007. https://doi.org/10.1080/22221751.2020.1756698.
Yan C, Cui J, Huang L, Du B, Chen L, Xue G, et al. Rapid and visual detection of 2019 novel coronavirus (SARS-CoV-2) by a reverse transcription loop-mediated isothermal amplification assay. Clin Microbiol Infect. 2020;6:773–9. https://doi.org/10.1016/j.cmi.2020.04.001.
Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat Biotechnol. 2020;7:870–4. https://doi.org/10.1038/s41587-020-0513-4.
Chan JF-W, Yip CC-Y, To KK-W, Tang TH-C, Wong SC-Y, Leung K-H, et al. Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens. J Clin Microbiol. 2020;5:e00310–20. https://doi.org/10.1128/JCM.00310-20.
**e C, Jiang L, Huang G, Pu H, Gong B, Lin H, et al. Comparison of different samples for 2019 novel coronavirus detection by nucleic acid amplification tests. Int J Infect Dis. 2020:264–7. https://doi.org/10.1016/j.ijid.2020.02.050.
Ai T, Yang Z, Hou H, Zhan C, Chen C, Lv W, et al. Correlation of chest CT and RT-PCR testing for coronavirus disease 2019 (COVID-19) in China: a report of 1014 cases. Radiology. 2020;2:E32–40. https://doi.org/10.1148/radiol.2020200642.
Afzal A. Molecular diagnostic technologies for COVID-19: limitations and challenges. J Adv Res. 2020;26:149–59. https://doi.org/10.1016/j.jare.2020.08.002.
Gibani MM, Toumazou C, Sohbati M, Sahoo R, Karvela M, Hon T-K, et al. Assessing a novel, lab-free, point-of-care test for SARS-CoV-2 (CovidNudge): a diagnostic accuracy study. Lancet Microbe. 2020. https://doi.org/10.1016/S2666-5247(20)30121-X.
D'Cruz RJ, Currier AW, Sampson VB. Laboratory testing methods for novel severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2). Front Cell Dev Biol. 2020;468. https://doi.org/10.3389/fcell.2020.00468.
Rabe BA, Cepko C. SARS-CoV-2 detection using isothermal amplification and a rapid, inexpensive protocol for sample inactivation and purification. Proc Natl Acad Sci. 2020;39:24450. https://doi.org/10.1073/pnas.2011221117.
Vandenberg O, Martiny D, Rochas O, van Belkum A, Kozlakidis Z. Considerations for diagnostic COVID-19 tests. Nat Rev Microbiol. 2020. https://doi.org/10.1038/s41579-020-00461-z.
Zeng H, Deng S, Zhou Z, Qiu X, Jia X, Li Z, et al. Diagnostic value of combined nucleic acid and antibody detection in suspected COVID-19 cases. Public Health. 2020:1–5. https://doi.org/10.1016/j.puhe.2020.07.011.
Ma H, Zeng W, He H, Zhao D, Jiang D, Zhou P, et al. Serum IgA, IgM, and IgG responses in COVID-19. Cell Mol Immunol. 2020;7:773–5. https://doi.org/10.1038/s41423-020-0474-z.
**ang F, Wang X, He X, Peng Z, Yang B, Zhang J, et al. Antibody detection and dynamic characteristics in patients with coronavirus disease 2019. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa461.
Van Elslande J, Houben E, Depypere M, Brackenier A, Desmet S, André E, et al. Diagnostic performance of seven rapid IgG/IgM antibody tests and the Euroimmun IgA/IgG ELISA in COVID-19 patients. Clin Microbiol Infect. 2020;8:1082–7. https://doi.org/10.1016/j.cmi.2020.05.023.
GeurtsvanKessel CH, Okba NMA, Igloi Z, Bogers S, Embregts CWE, Laksono BM, et al. An evaluation of COVID-19 serological assays informs future diagnostics and exposure assessment. Nat Commun. 2020;1:3436. https://doi.org/10.1038/s41467-020-17317-y.
Nicol T, Lefeuvre C, Serri O, Pivert A, Joubaud F, Dubée V, et al. Assessment of SARS-CoV-2 serological tests for the diagnosis of COVID-19 through the evaluation of three immunoassays: two automated immunoassays (Euroimmun and Abbott) and one rapid lateral flow immunoassay (NG Biotech). J Clin Virol. 2020;104511. https://doi.org/10.1016/j.jcv.2020.104511.
Wu J-L, Tseng W-P, Lin C-H, Lee T-F, Chung M-Y, Huang C-H, et al. Four point-of-care lateral flow immunoassays for diagnosis of COVID-19 and for assessing dynamics of antibody responses to SARS-CoV-2. J Inf Secur. 2020;3:435–42. https://doi.org/10.1016/j.**f.2020.06.023.
Carter LJ, Garner LV, Smoot JW, Li Y, Zhou Q, Saveson CJ, et al. Assay techniques and test development for COVID-19 diagnosis. ACS Cent Sci. 2020;5:591–605. https://doi.org/10.1021/acscentsci.0c00501.
Liu W, Liu L, Kou G, Zheng Y, Ding Y, Ni W, et al. Evaluation of nucleocapsid and spike protein-based enzyme-linked immunosorbent assays for detecting antibodies against SARS-CoV-2. J Clin Microbiol. 2020;6:e00461–20. https://doi.org/10.1128/JCM.00461-20.
Chen Z, Zhang Z, Zhai X, Li Y, Lin L, Zhao H, et al. Rapid and sensitive detection of anti-SARS-CoV-2 IgG, using lanthanide-doped nanoparticles-based lateral flow immunoassay. Anal Chem. 2020;10:7226–31. https://doi.org/10.1021/acs.analchem.0c00784.
Wan WY, Lim SH, Seng EH. Cross-reaction of sera from COVID-19 patients with SARS-CoV assays. medRxiv. 2020. https://doi.org/10.1101/2020.03.17.20034454.
Tay MZ, Poh CM, Rénia L, MacAry PA. Ng LFP the trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;6:363–74. https://doi.org/10.1038/s41577-020-0311-8.
Su B, Yin J, Lin X, Zhang T, Yao X, Xu Y, et al. Quantification of SARS-CoV-2 antigen levels in the blood of patients with COVID-19. Sci China Life Sci. 2020. https://doi.org/10.1007/s11427-020-1830-8.
Norman M, Gilboa T, Ogata AF, Maley AM, Cohen L, Busch EL, et al. Ultrasensitive high-resolution profiling of early seroconversion in patients with COVID-19. Nat Biomed Eng. 2020;12:1180–7. https://doi.org/10.1038/s41551-020-00611-x.
Li Y-H, Li J, Liu X-E, Wang L, Li T, Zhou Y-H, et al. Detection of the nucleocapsid protein of severe acute respiratory syndrome coronavirus in serum: comparison with results of other viral markers. J Virol Methods. 2005;1:45–50. https://doi.org/10.1016/j.jviromet.2005.06.001.
Kammila S, Das D, Bhatnagar PK, Sunwoo HH, Zayas-Zamora G, King M, et al. A rapid point of care immunoswab assay for SARS-CoV detection. J Virol Methods. 2008;1:77–84. https://doi.org/10.1016/j.jviromet.2008.05.023.
Shan D, Johnson JM, Fernandes SC, Suib H, Hwang S, Wuelfing D, et al. N-protein presents early in blood, dried blood and saliva during asymptomatic and symptomatic SARS-CoV-2 infection. Nat Commun. 2021;1:1931. https://doi.org/10.1038/s41467-021-22072-9.
Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;6:595–9. https://doi.org/10.1038/nbt.1641.
Cohen L, Walt DR. Highly sensitive and multiplexed protein measurements. Chem Rev. 2019;1:293–321. https://doi.org/10.1021/acs.chemrev.8b00257.
Chen Y, Cai Q, Liu S. A single molecule assay for ultrasensitive detection of Fn14 in human serum. Anal Biochem. 2019;587, 113467. https://doi.org/10.1016/j.ab.2019.113467.
Rissin DM, Fournier DR, Piech T, Kan CW, Campbell TG, Song L, et al. Simultaneous detection of single molecules and singulated ensembles of molecules enables immunoassays with broad dynamic range. Anal Chem. 2011;6:2279–85. https://doi.org/10.1021/ac103161b.
Kan CW, Rivnak AJ, Campbell TG, Piech T, Rissin DM, Mösl M, et al. Isolation and detection of single molecules on paramagnetic beads using sequential fluid flows in microfabricated polymer array assemblies. Lab Chip. 2012;5:977–85. https://doi.org/10.1039/C2LC20744C.
Funding
This work was supported by the National Natural Science Foundation of China (21974125), the Foundation for University Key Teacher by Henan Province (2017GGJS007), the National 111 project (D20003), and the Key Scientific Research Project in Universities of Henan Province (19A150048).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics declarations
The human serum samples were collected at the First Affiliated Hospital of Zhengzhou University. All the experiments in this work were carried out in compliance with ethical standards, and conducted according to the Declaration of Helsinki and approved by the Ethics Committee of Zhengzhou University.
Conflict of interest
The authors declare no competing interests.
Additional information
Published in the topical collection Analytical Chemistry for Infectious Disease Detection and Prevention with guest editors Chaoyong Yang and **uJun (James) Li.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 276 kb)
Rights and permissions
About this article

Cite this article
Cai, Q., Mu, J., Lei, Y. et al. Simultaneous detection of the spike and nucleocapsid proteins from SARS-CoV-2 based on ultrasensitive single molecule assays. Anal Bioanal Chem 413, 4645–4654 (2021). https://doi.org/10.1007/s00216-021-03435-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03435-z