
Abstract
Key message
An epistatic interaction between SCN resistance loci rhg1-a and rhg2 in PI 90763 imparts resistance against virulent SCN populations which can be employed to diversify SCN resistance in soybean cultivars.
Abstract
With more than 95% of the $46.1B soybean market dominated by a single type of genetic resistance, breeding for soybean cyst nematode (SCN)-resistant soybean that can effectively combat the widespread increase in virulent SCN populations presents a significant challenge. Rhg genes (for Resistance to Heterodera glycines) play a key role in resistance to SCN; however, their deployment beyond the use of the rhg1-b allele has been limited. In this study, quantitative trait loci (QTL) were mapped using PI 90763 through two biparental F3:4 recombinant inbred line (RIL) populations segregating for rhg1-a and rhg1-b alleles against a SCN HG type 1.2.5.7 (Race 2) population. QTL located on chromosome 18 (rhg1-a) and chromosome 11 (rhg2) were determined to confer SCN resistance in PI 90763. The rhg2 gene was fine-mapped to a 169-Kbp region pinpointing GmSNAP11 as the strongest candidate gene. We demonstrated a unique epistatic interaction between rhg1-a and rhg2 loci that not only confers resistance to multiple virulent SCN populations. Further, we showed that pyramiding rhg2 with the conventional mode of resistance, rhg1-b, is ineffective against these virulent SCN populations. This highlights the importance of pyramiding rhg1-a and rhg2 to maximize the impact of gene pyramiding strategies toward management of SCN populations virulent on rhg1-b sources of resistance. Our results lay the foundation for the next generation of soybean resistance breeding to combat the number one pathogen of soybean.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean cyst nematode (SCN; Heterodera glycines, Ichinohe) is a major threat to soybean production worldwide (Koenning and Wrather 2010; Allen et al. 2017; Tylka and Marett 2021). The monetary loss for soybean producers caused by SCN from 1996 through 2016 is estimated to be $32 billion with more than $1.5 billion in yield losses annually in the USA (Bandara et al. 2020). Current SCN management practices are based on an approach that includes planting-resistant cultivars, utilizing seed treatments, and implementing non-host crop rotations (Concibido et al. 2004; Niblack 2005; Mitchum 2016). Although plant genetic resistance is the most cost-effective and reliable management strategy, the genetic complexity of resistance poses a major barrier for breeding SCN resistance into new soybean cultivars (Niblack et al. 2008; Mitchum 2016). SCN management issues are further exacerbated by the existence of SCN populations with diverse virulence profiles (Niblack et al. 2002, 2008; Concibido et al. 2004; Tylka 2016). Therefore, a more complete understanding of known plant genetic resistance against modern virulent SCN populations is crucial for sustainable SCN management.
The pioneering discovery of Rhg genes (for Resistance to H. glycines) dates to the 1960s (Caldwell et al. 1960). Since then, there has been tremendous progress in understanding SCN genetic resistance through map** of quantitative trait loci (QTL) from diverse soybean germplasm and the molecular characterization of some of these genes (Concibido et al. 2004; Mitchum 2016). Two major SCN resistance loci cqSCN-001 (Rhg1) and cqSCN-002 (Rhg4) were commonly mapped in different soybean germplasm and extensively utilized in the development of modern SCN-resistant soybean cultivars (Concibido et al. 2004; Liu et al. 2012, 2017; Cook et al. 2012, 2014; Mitchum 2016; Bayless et al. 2019). The classification of SCN resistance sources into plant introduction (PI) 88788 and Peking types was also primarily based on the presence of these two loci and their allelic variants (Brucker et al. 2005; Bayless et al. 2019). The rhg1-b resistance to SCN HG type 0 (Race 3) in PI 88788 is governed by a 31-kb repeated genomic region containing GmSNAP18 (α-soluble N-ethylmaleimide sensitive factor attachment protein), one of the three genes contributing to SCN resistance (Cook et al. 2012, 2014; Lee et al. 2015). An epistatic interaction between the rhg1-a SNAP18 allele and the Rhg4 locus encoding a serine hydroxymethyltransferase (SHMT08) governs bi-genic resistance to SCN HG type 0 (Race 3) in Peking types such as the cultivar Forrest (Liu et al. 2012, 2017; Kandoth et al. 2017). However, these two resistance loci alone are incapable of explaining resistance mechanisms in all diverse soybean germplasm known to exhibit SCN resistance.
PI 88788 has been excessively utilized as a resistance source in modern SCN-resistant soybean cultivars due to resistance being derived from a single rhg1-b allele which allows for convenient breeding by introgression of a single resistance locus (Concibido et al. 2004; Niblack, 2005; Mitchum 2016; McCarville et al. 2017). Consequently, the monoculture of PI 88788-resistant soybean cultivars has facilitated the selection of virulent nematode populations that are capable of overcoming this resistance (Niblack et al. 2008; McCarville et al. 2017; Howland et al. 2018; Meinhardt et al. 2021). A crucial step to a successful SCN management strategy is the use of resistance sources capable of limiting the selection of virulent nematode populations which has often been undermined while breeding for high-yielding SCN-resistant cultivars (Niblack 2005; Chen 2020; Meinhardt et al. 2021). Due to the limited understanding of SCN virulence genes referred to as ror genes (for the reproduction on a resistant host), a strategic rotation of SCN resistance sources that can counter-select nematode populations is a potential solution for combating SCN (Gardner et al. 2017; Meinhardt et al. 2021). Counter-selection studies using different virulent nematode populations on PI 88788, PI 90763, and Peking have contributed to insights on the potential use of these sources in rotation to limit virulent SCN population build-up (Luedders and Dropkin 1983; Anand and Shumway 1985; Gardner et al. 2017; Meinhardt et al. 2021). Some of these studies have highlighted a strong counter-selection between virulence on PI 88788 and PI 90763 (Anand and Shumway 1985; Gardner et al. 2017). This has further elevated the unique resistance in PI 90763, which has been underutilized in traditional and modern cultivar development programs.
Several genetic map** studies have reported approximately 216 SCN-resistant QTL from multiple sources on different chromosomes (SoyBase 2021). Confirmed SCN resistance QTL include cqSCN-001, cqSCN-002, cqSCN-003, cqSCN-005, cqSCN-006, and cqSCN-007 (SoyBase 2021). The QTL cqSCN-001 and cqSCN-002 correspond to the Rhg1 and Rhg4 loci, respectively. Other SCN resistance QTL were mapped and confirmed, from multiple PIs and using multiple SCN populations, on Chr. 10 as cqSCN10 (Vuong et al. 2010; Zhou et al. 2021), 11 as cqSCN11/rhg2 (Wu et al. 2009; St. Amour et al. 2020; Suzuki et al. 2020), 15 as cqSCN-006 (Kim and Diers 2013; Yu and Diers 2017), 16 as cqSCN-003 (Concibido et al. 1997; Glover et al. 2004), 17 as cqSCN-005 (Kazi et al. 2010), and 18 as cqSCN-007 (Vuong et al. 2010; Kim and Diers 2013; Yu and Diers 2017; Usovsky et al. 2021a). PI 90763 is a highly resistant source against SCN populations with diverse virulent profiles (Anand and Shumway 1985; Arelli et al. 1997; Guo et al. 2005; Gardner et al. 2017). It has also been utilized as an indicator line in the SCN (HG) type test (Niblack et al. 2002). SCN resistance in PI 90763 to multiple SCN populations has been mapped to Chr. 18 by Concibido et al. (1997) using restriction fragment length polymorphism (RFLP) markers. Later, Guo et al. (2005) reported QTL on Chr. 6, 8, 11, 15, 16, 18, and 19 using simple sequence repeat (SSR) markers and multiple SCN populations. However, limited research has been conducted to determine the impact of different known SCN resistance loci from PI 90763 and their interactions with rhg1-b. SCN resistance in PI 90763 is underutilized with limited resistance map** studies and no reports on the incorporation of SCN resistance loci from PI 90763 into modern resistant cultivars to date (Kofsky et al. 2021). The fundamental reason behind the underutilization of the PI 90763 source is potentially attributed to its similar resistance response as Peking-type sources (Myers and Anand 1991; Concibido et al. 1997; Guo et al. 2005). Hence, the genetic resistance in PI 90763 can be a useful resource in unraveling different resistance mechanisms among Peking-type sources and decoding counter-selection of SCN virulence genes, which could aid in the design of a long-term SCN management strategy. Thus, the specific objectives of this study were to: (1) determine genomic regions governing SCN resistance in PI 90763 to SCN HG type 1.2.5.7 through linkage and nested association map** (NAM) strategies, (2) fine map the rhg2 gene to identify potential candidate genes, and (3) investigate the impact of resistance allele combinations against different SCN populations to devise an effective long-term SCN management strategy.
Materials and methods
Population development
To study SCN resistance in PI 90763 through genetic linkage analysis, we developed two F3:4 recombinant inbred line (RIL) map** populations by crossing resistant parents with contrasting rhg1-a and rhg1-b alleles at the Rhg1 locus. The usefulness of F3-derived populations in genetic map** studies has been previously established (Takuno et al. 2012). The first map** population (pop1) was developed by crossing high-yielding elite soybean line SA13-1385 (rhg1-b; PI 88788 type) with PI 90763 (rhg1-a). Similarly, the second map** population (pop2) was created by crossing the high-yielding elite soybean line LD11-2170 (rhg1-b; PI 88788 type) and PI 90763 (rhg1-a). A third population developed by a cross between a susceptible soybean line SA10-8471 (pop3) and PI 90763 (rhg1-a) was used along with pop1 and pop2 for the NAM analysis.
PI 90763 is highly resistant to multiple SCN populations (Arelli et al. 1997; Klepadlo et al. 2018), whereas SA13-1385 and LD11-2170 carry PI 88788-type resistance and were released as SCN-resistant varieties from the University of Missouri-Columbia and the University of Illinois, Urbana-Champaign, respectively. SA13-1385 and LD11-2170 are highly resistant to HG type 0. A total of 330, 274, and 218 RILs were developed for pop1, pop2, and pop3, respectively. The cross-pollination for pop1 and pop2 were made at the Bay Farm Research Facility in Columbia, MO during the summer of 2017. The RIL populations were inbred and advanced using the single-seed descent method (Brim 1966) at Hartung Brothers Inc. winter nursery in Kekaha, HI. Hybrid F1 seeds were harvested during the summer of 2017 and were sent to Kauai, Hawaii, for advancing generations at the winter nursery in October 2017. The cross-pollination for pop 3 was made at the Bay Farm Research Facility in Columbia, MO, during the summer of 2019, and F3:4 RILs from pop3 were similarly developed.
SCN bioassay
The soybean cyst nematode bioassay was conducted following the standardized cyst evaluation protocol (Niblack et al. 2009). The SCN inbred population TN22, HG type 1.2.5.7 (Race 2) was used as the inoculum source. Five seedlings from each F3:4 line were transplanted along with parental lines, the susceptible checks Lee 74 and Williams 82, Pickett, and the HG type test indicator lines (PI 548402, PI 88788, PI 90763, PI 437654, PI 209332, PI 89772, and PI 548316) (Niblack et al. 2002). Each map** population was tested at a different period due to limited greenhouse capacity. The seedlings were inoculated with 1200 eggs two days after transplanting. Each of five replicated seedlings per experimental line was planted in a different micro-pot and organized in a randomized complete block design. A root temperature of 27 °C was maintained throughout the experiment. Thirty days after inoculation, roots were soaked in water to remove the soil and sprayed with high-pressure water over a set of nested sieves (no. 20 over no. 60). The cysts obtained from each plant root system were manually counted under a stereoscope, and an average number of cysts for each line was determined. Female indices (FI) were calculated for each line by dividing the average number of cysts on each line by the average number of cysts on the susceptible parent and multiplying by 100 (Nibalck et al. 2002). Experimental lines were rated following a standardized method as resistant (R, FI < 10%), moderately resistant (MR, FI = 10–30%), moderately susceptible (MS, FI = 31–60%), and susceptible (S, FI > 60%) (Schmitt and Shannon 1992). Shapiro–Wilk's test was performed in RStudio to determine the normality of distribution of female indices while symmetry was analyzed using skewness and kurtosis of the distributions.
Genoty** and SNP analysis
Genomic DNA was extracted with the cetyl trimethyl ammonium bromide (CTAB) method (Doyle and Doyle 1987). 10 young trifoliate leaves from 10 to 15 plants of each F3:4 line were bulked, freeze-dried and DNA samples were extracted. The extracted DNA samples from the three populations were submitted to the Soybean Genomics and Improvement Laboratory, USDA-ARS, Beltsville, MD, for single-nucleotide polymorphism (SNP) genoty** using the Illumina Infinium BARCSoySNP6K BeadChip (Song et al. 2020), and allele calls were made using Genome Studio software (Illumina Inc). SNP markers obtained from genoty** using the Illumina Infinium BARCSoySNP6K BeadChip for both populations were filtered using TASSEL software (Bradbury et al. 2007). The RILs exceeding more than 10% of missing calls and 30% heterozygosity were eliminated. The SNP matrix was then converted to A/B/H format for both the populations individually using ABH genotype in TASSEL (Bradbury et al. 2007). The ABHgenotypeR package was used in RStudio to conduct an imputation of the missing genotypes based on flanking alleles (Reuscher and Furuta 2016). A similarity test was conducted to eliminate RILs below 90% genetic similarity between the parents using the R package “ParentOffSpring” (Abdel-Haleem et al. 2013). Obtained SNP matrixes were utilized for constructing genetic linkage maps using the qtl package in RStudio (Browman and Sen 2009).
Genetic linkage and nested association map**
Map** of quantitative trait loci was performed using MapQTL 5.0 (van Ooijen 2004). Interval map** (IM) at 1-cM intervals along the chromosome was used to detect QTL based on a LOD threshold of 3.0. This corresponds to a false discovery rate (FDR) of 0.05, which was determined by permutation tests. Markers closely linked to positions with the highest LOD scores were taken as cofactors for multiple-QTL modeling (MQM) analysis (van Ooijen 2004). Graphical presentation of QTL was drawn using MapChart 2.3 (Voorrips 2002). The composite interval map** (CIM) through the RStudio qtl package was performed for the further confirmation of QTL map** results obtained from the MapQTL (Browman and Sen 2009).
Nested Association Map** was conducted through the NAM package in RStudio with an efficient mixed-model association algorithm (Xavier et al. 2015) using the three F3:4 RIL populations for NAM analysis. To determine the significance of SNPs in the association map**, an FDR threshold at α ≤ 0.001 was calculated, and SNPs were declared significant based on the FDR threshold value.
Fine-map** of rhg2 locus
Twenty-four F3:5 lines homozygous at rhg1-a and heterozygous at the rhg2 from pop1 were selected based on confidence intervals delimited in this study and confirmed using two KASP assays (Kompetitive Allele Specific PCR): Rhg1-2 and SNAP11 (Kadam et al. 2016; Usovsky et al. 2021a). The presence/absence of the Rhg4 resistance allele was confirmed by KASP assay using Rhg4-5 marker (Kadam et al. 2016). Twenty plants from each of the selected lines were tagged individually, young trifoliate leaves were collected and freeze-dried, and DNA was extracted following the modified CTAB method as previously described. Four KASP assays (Rhg1-2, SNAP18-1, SNAP11-1, and Rhg4-5) were used to confirm the homozygosity state of each F5:6 plant. A set of 24 KASP assays (named as “MU”) were newly designed based on the whole-genome sequencing data to detect the recombination spots near the rhg2 locus (www.SoyBase.org). The F5:6 lines were phenotyped with SCN population TN22 (HG type 1.2.5.7) with five replications following standardized procedures (Niblack et al. 2009).
SCN screening of allelic combinations
Ninety-two F3:5 lines from pop1 homozygous at Rhg1, rhg2, and Rhg4 were selected based on confidence intervals delimited in this study, which was confirmed using four Kompetitive Allele Specific PCR (KASP) assays: Rhg1-2 (For rhg1), SNAP18-1 (For GmSNAP18), SNAP11-1 (For GmSNAP11, and Rhg4-5 (For Rhg4) (Kadam et al. 2016; Usovsky et al. 2021a). These lines were separated into eight different categories based on their allelic combinations: A1 = rhg1-a (n = 15), A2 = rhg1-a + rhg2 (n = 10), A3 = rhg1-a + rhg2 + Rhg4 (n = 5), A4 = rhg1-a + Rhg4 (n = 21), B1 = rhg1-b (n = 4), B2 = rhg1-b + rhg2 (n = 17), B3 = rhg1-b + rhg2 + Rhg4 (n = 15), and B4 = rhg1-b + Rhg4 (n = 5). Similarly, 69 F3:5 lines were selected from pop2 where only four of the above allelic combinations were present as rhg2 was not segregating. These four allelic combinations included: A2 = rhg1-a + rhg2 (n = 14), A3 = rhg1-a + rhg2 + Rhg4 (n = 16), B2 = rhg1-b + rhg2 (n = 15), and B3 = rhg1-b + rhg2 + Rhg4 (n = 24).
Selected RILs with different allelic combinations for pop1 and pop2 were screened against SCN inbred populations TN7 (HG type 2.5.7; race 1), TN22 (HG type 1.2.5.7; race 2), PA3 (HG type 0; race 3), and MM4 (HG type 2.5.7; race 5). Screening of selected RILs in pop1 and pop2 was conducted at two different periods with five replications in a completely randomized design. SCN inoculations and data collection were conducted based on the standardized screening procedure as previously described. Female indices calculated from the screening of the allelic combinations from both populations were analyzed by one-way analysis of variance (ANOVA) in RStudio, and Tukey’s HSD test was used for multiple comparisons of SCN phenotypes with different allelic combinations.
Results
Phenotypic variation of SCN resistance and genetic linkage map**
Two biparental map** populations were screened against the TN22 SCN population to map resistance QTL regions. Additionally, seven HG type indicator lines, two susceptible lines, and one race differential line confirmed the correct responses of HG type 1.2.5.7 (race 2) (Supplemental Table 1). The FI of parental lines for pop1 was 83 and 0 for SA13-1385 and PI 90763, respectively (Fig. 1A; Table 1). For pop2, the FI of parental lines LD11-2170 and PI 90763 were 94 and 0, respectively (Fig. 1B; Table 1). Therefore, both of the parents SA13-1385 and LD11-2170 were highly susceptible and PI 90763 was highly resistant against SCN population TN22. The phenotypic distribution of FI in pop1 ranged from 0 to 167 with a mean of 106 based on the susceptible parent SA13-1385 (Fig. 1a; Table 1), whereas the phenotypic distribution of FI in pop2 ranged from 0 to 176 with a mean of 80 based on the susceptible parent LD11-2170 (Fig. 1b; Table 1). The differences in frequency distributions indicated divergent genetic backgrounds for SCN resistance in pop1 and pop2.

Frequency distribution of female indices against SCN population TN22 (HG type 1.2.5.7). A 303 F3:4 RILs from pop1 (SA13-1385 × PI 90763) and B 251 F3:4 RILs from pop2 (LD11-2170 × PI 90763)
Genetic linkage maps for both populations were constructed using the qtl package in RStudio based on the segregation of SNP markers across 20 chromosomes. Approximately 6000 raw SNPs were obtained from the Illumina Infinium SoySNP6K assay for each population. The number of high-quality polymorphic markers showing distinct segregation that were obtained after filtering was 2265 for pop1 and 2123 for pop2 (Fig. 2A, B; Table 2). SNP markers spanned through 4177.7 and 4090.3 cM for pop1 and pop2, respectively, and both linkage maps provided comparable coverage of SNPs in both populations (Table 2; Fig. 2). Two major QTL on Chr. 11 and 18 were detected by the multiple-QTL modeling (MQM) method in pop1 (Fig. 3A; Table 3). The QTL on Chr. 11 and 18 explained 28.3 and 23.9% of total phenotypic variance and their additive effects were 23.2 and 21.5, respectively, and PI 90763 was the source of resistance alleles for both. The Chr. 11 QTL was mapped to a 575-kbp interval between the markers Gm11_37237023—Gm11_37749863 (Wm82.a2. v1) with a peak at Gm11_37408299 marker (Gm11:32959788; Wm82.a2. v1). The Chr. 18 QTL was mapped to a 374-kbp interval between the markers Gm18_1562162 and Gm18_1909453 (Wm82.a2. v1) with a peak at Gm18_1909453 marker (Gm18:1909982; Wm82.a2. v1) (Fig. 3, Table 3). CIM analysis performed in RStudio using the qtl package detected both QTL along with their significant epistatic interaction (Supplemental Fig. 1A; Supplemental Table 2; 3A).

Genetic linkage map created for A 303 F3:4 RILs from pop1 (SA13-1385 and PI 90763) and B 251 F3:4 RILs from pop2 (LD11-2170 × PI 90763). The X-axis represents chromosome numbers, and Y-axis represents the genetic position of single-nucleotide polymorphism (SNP) markers. Distribution and SNPs are represented by black bars across each chromosome

Quantitative trait loci (QTL) controlling soybean cyst nematode (SCN) resistance to TN22 (HG type 1.2.5.7) in PI 90763: A two major QTL detected in pop1 (SA13-1385 × PI 90763); B one major QTL detected in pop2 (LD11-2170 × PI 90763). Scales on the left of the chromosome represent the map position in centiMorgans (cM). Scales on the top of the graph represent the value of the logarithm of the odds (LOD). The black dotted line indicates the threshold of significance (LOD = 3.5 and 3.4) for pop1 and pop2, respectively
A single major QTL was mapped to Chr. 18 using the MQM method in pop2 which explained 76.4% of the total phenotypic variation (Fig. 3B; Table 3). The additive effect for the QTL was 50.1 with a resistance allele originating from PI 90763 (Table 3). The QTL spanned through the 1080-Kbp region where the confidence interval was established between the markers Gm18_829765 and Gm18_1909453 that corresponded to a physical location of Gm18:830106–1909982 (Wm82.a2.v1) with a peak at Gm18_1562162 marker (Gm18:1562536; Wm82.a2.v1) (Fig. 3B; Table 3). CIM analysis performed using qtl package in RStudio detected the same QTL (Supplemental Fig. 1B; Supplemental Table 2; 3B). The genomic regions of Chr. 18 in both the map** populations correspond to the Rhg1 locus, whereas the rhg2 gene nomenclature is now associated with the genomic region of the QTL on Chr. 11. Although rhg2 has been detected in a few other resistance sources, the underlying gene(s) have not been cloned. Hence, the genetic linkage analysis revealed rhg1-a and rhg2 resistance loci govern resistance against the SCN inbred population TN22 (HG type 1.2.5.7).
Nested association map**
SNP markers with the greatest LOD score for resistance to the TN22 population (HG type 1.2.5.7) were identified in the NAM analysis. Additive allelic effects were determined relative to the hub parent PI 90763 where a positive effect represented an increase in FI due to allele substitution of PI 90763 alleles by alleles from the founder parents. A negative effect represented a reduction in FI due to allele substitution of founder parent allele with an allele from PI 90763. Using a false discovery rate threshold of α ≤ 0.001, several SNPs associated with SCN resistance were identified from the NAM analysis (Table 4) and these mapped to the Chr. 11 and 18 intervals identified by the MQM analysis. However, the significance of QTL detected through the NAM population in Chr. 11 was lower than that in pop1 using the MQM analysis (Fig. 4; Supplementary Fig. 1A). This depicts that the allele frequency in map** populations primarily contributes to the significance of QTL in NAM analysis (Yu et al. 2008; Beche et al. 2020). The NAM analysis further detected an additional QTL on Chr. 03 using three map** populations (Fig. 4; Table 4), whereas only two QTL peaks were detected in pop3 in Chr.11 and 18 (data not shown) through the linkage analysis. These data further corroborate the power of rare allele detection in the NAM analysis as observed with increased detection of trait-associated markers (Yu et al. 2008; Beche et al. 2020). The top three significant SNPs for each QTL obtained from the NAM analysis are listed in Table 4. SCN resistance QTL on Chr. 03 was previously determined for HG type 2.5.7 (race 1) in PI 90763 and HG type 0 (race 6) in PI 209332 (Concibido et al. 1997), HG type 2.5.7 (race 5) on PI 404198A (Guo et al. 2006), HG type 1.3.5.6.7 and HG type 1.2.5.7 in PI 437655 (Jiao et al. 2015). Thus, the NAM analysis reaffirmed the QTL obtained from the linkage map** analysis along with an additional QTL on Chr. 03 against SCN population TN22 (HG type 1.2.5.7).

Manhattan plots of nested association map** (NAM) analysis for SCN population TN22 (HG type 1.2.5.7) plotted against positions on each of the 20 chromosomes. The NAM panel included pop1 (SA13-1385 × PI 90763), pop2 (LD11-2170 × PI 90763) and pop3 (SA10-8471 × PI 90763). The significant SNPs were distinguished by the false discovery rate (FDR) of the α ≤ 0.001 thresholds represented by the dotted line
Fine-map** of rhg2 gene
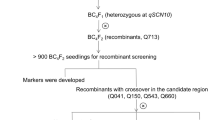
Fine-map** of the rhg2 gene was conducted to narrow the genetic region and pinpoint candidate genes imparting SCN resistance. Twenty F3:5 RILs from pop1 were pooled from RILs with both the loci where rhg1-a was homozygous and rhg2 was homozygous at one end and heterozygous at the other end of the confidence interval determined by QTL map** (Table 3). These lines were advanced to create F5:6 sister lines for each recombination event within the rhg2 region. Six crossing-over events were identified in lines SA18-17394, SA18-17486, SA18-17447, SA18-17176, SA18-17229, and SA18-17370 (Fig. 5). The fine-map** analysis further delimited the rhg2 gene between MU-35 and MU-52 markers indicating that rhg2 is in the 169-Kbp interval between MU-35 and MU-52 (Gm11:32906157–33075108; Wm82. a.2.v.1) (Supplemental Table 4). There are 21 potential candidate genes within the fine-mapped region based on the Williams 82 reference genome (www.SoyBase.org). The strongest candidate was found to be GmSNAP11 gene, a paralog of the GmSNAP18 gene at the Rhg1 locus (Cook et al. 2012, p. 14; Lakhssassi et al. 2017; St-Amour et al. 2020; Usovsky et al. 2021b). This result further suggests that PI 88788-type GmSNAP18, Peking-type GmSNAP18, and their paralog GmSNAP11 have evolved to underlie different types of resistance and/or diversified function of a pleiotropic role of Peking-type GmSNAP18 and GmSNAP11 as in reniform nematode resistance (Usovsky et al. 2021b). Our map** of the rhg2 locus to a 169-Kbp interval is smaller than the 821-Kbp region; it was previously mapped to (Suzuki et al. 2020). Hence, the rhg2 gene was fine-mapped to 169-Kbp region and the GmSNAP18 gene was identified as the potential candidate gene.

Phenotypic responses of lines carrying eight allelic combinations from pop1 (SA13-1385 × PI 90763) tested against SCN populations a TN7 (HG type 2.5.7; race 1), b TN22 (HG type 1.2.5.7; race 2), c PA3 (HG type 0; race 3), and d MM4 (HG type 2.5.7; race 5). The X-axis represents different allelic combinations of homozygous SCN resistance alleles/QTL. The numbers and letters above each box plot indicate the number of lines tested and significance grou** based on Tukey's honest significant difference test (Tukey's HSD) at P ≤ 0.05. Legend: A1 = rhg1-a; A2 = rhg1-a + rhg2; A3 = rhg1-a + rhg2 + Rhg4; A4 = rhg1-a + Rhg4, B1 = rhg1-b; B2 = rhg1-b + rhg2; B3 = rhg1-b + rhg2 + Rhg4; B4 = rhg1-b + Rhg4
SCN screening of specific allelic combinations of rhg1-a, rhg1-b, rhg2, and Rhg4 loci
SCN screening of different Rhg combinations demonstrated their impact against different SCN populations. Subgroups with rhg1-a + rhg2 + Rhg4 (FI = 0.6; Tukey’s HSD mean separation (MS) = a) and rhg1-a + rhg2 (FI = 13, MS = a) in pop1 had similar FI and were the most resistant among all the combinations for TN7 (HG type 2.5.7) (Fig. 6A). Similar SCN phenotypic responses were observed for TN22 (HG type 1.2.5.7) and MM4 (HG type 2.5.7) SCN populations for rhg1-a + rhg2 + Rhg4 (FI = 11, MS = a; FI = 1, MS a) and rhg1-a + rhg2 (FI = 8, MS = a; FI = 19, MS = b), respectively (Fig. 6B, D). The SCN phenotypic responses were validated by screening resistance loci combinations from pop2 which resulted in similar responses observed previously for pop1 (Fig. 7). The results confirmed the epistatic interaction of the rhg1-a and rhg2 loci contributing resistance against TN7, TN22, and MM4. Consistent with prior studies, either rhg1-b alone or a combination of rhg1-a and Rhg4 was sufficient for resistance to SCN HG type 0 (Race 3) (Figs. 6C, 7). The other combinations including the rhg1-b allele displayed moderately susceptible or susceptible phenotypic responses against the three SCN populations (Fig. 6a, b, d). Thus, the results highlight that a pyramid of the rhg1-a allele with rhg2 offers an advantage against the virulent SCN populations tested. Since rhg1-b is the most widely used resistance allele in more than 95% of resistant soybean cultivars planted in the USA, current efforts have focused on pyramiding of rhg1-b with other resistance loci to enhance resistance to virulent SCN (Brzostowski and Diers 2017; Yu and Diers 2017; Meinhardt et al. 2021). Here, we demonstrated that a combination of rhg1-a and rhg2 genes provides resistance against virulent nematode populations and resistance allele pyramid focused on these genes would be an effective strategy for the management of virulent SCN populations.

Phenotypic responses of lines carrying four allelic combinations from pop2 (LD11-2170 × PI 90763) tested against SCN populations A TN7(HG type 2.5.7; race 1), B TN22 (HG type 1.2.5.7; race 2), C PA3 (HG type 0; race 3), and D MM4 (HG type 2.5.7; race 5). The X-axis represents different allelic combinations of homozygous SCN resistance alleles/QTL. The numbers and letters above each box plot indicate the number of lines tested and significance grou** based on Tukey's test (Tukey's HSD) at P ≤ 0.05

Fine-map** of rhg2 gene in PI 90763. Alleles of Rhg1 locus (resistant Peking-type rhg1-a vs. PI 88788-type rhg1-b) and Rhg4 locus (resistant Peking-type Rhg4-a vs. susceptible Williams 82-type Rhg4-b) were confirmed using KASP assays (Kadam et al. 2016). Dark and light gray colors correspond to genomic regions derived from PI 90763 and SA13-1385, respectively. Vertical red lines signify recombination events between sister lines. Resistance rating to TN22 population (HG type 1.2.5.7) was calculated based on female index (FI): R = resistance (FI < 10), MR = moderate resistance (FI, 10–30), S = susceptibility (FI > 60)
Discussion
In this study, we demonstrated an epistatic interaction between rhg1-a and rhg2 loci in PI 90763, imparting SCN resistance against virulent SCN populations using two unique map** populations created by a cross between rhg1-b (SA13-1385/LD11-2170) and rhg1-a (PI 90763). We mapped rhg1-a and rhg2 in pop1, whereas only rhg1-a was mapped in pop2. Further KASP assays on parents determined that the rhg2 gene was present in the parent LD11-2170, which explained the detection of a single QTL on Chr. 18 in pop2. NAM analysis further confirmed the status of rhg2 allele frequency in NAM map** populations along with an additional QTL at Chr. 03. The significant rhg2 gene detected through genetic linkage and NAM analysis was further fine-mapped to a 169-kbp region using a genetic map** approach. GmSNAP11 was determined to be the strongest candidate gene. Previous reports on cloning of the Rhg1 locus in PI 88788 and Peking sources have characterized GmSNAP18 as a major gene imparting SCN resistance (Cook et al. 2012, 2014; Liu et al. 2017). The characterization of SNAP subfamily genes has demonstrated that the SNAP genes in soybean undergo co-regulation after SCN infection (Lakhssassi et al. 2017). The detection of SNAP genes at resistance loci rhg1-a, rhg1-b, and rhg2 highlights the crucial role of SNAPs in mediating SCN resistance against multiple virulent SCN populations and more recently reniform nematode (Lakhssassi et al. 2017; Usovsky et al. 2021b).
The epistatic interaction between rhg1-a and Rhg4 governs SCN HG type 0 (Race 3) resistance in Peking-type sources (Meksem et al. 2001; Concibido et al. 2004; Brucker et al. 2005; Liu et al. 2017). However, SCN HG type 0 (Race 3) resistant sources categorized as Peking type (Peking, PI 90763, PI 437654, and PI 89772) display distinct phenotypic responses against other SCN populations (Anand and Shumway 1985; Niblack 2005; Concibido et al. 2004; Niblack et al. 2006) that suggest a role for additional resistance genes. Efforts to determine the differential phenotypic responses within Peking type sources had been previously conducted by genetic analysis using map** populations derived from complementary crosses between Peking type sources (Thomas et al. 1975; Anand and Sharma 1995). However, an epistatic interaction of the rhg1-a and rhg2 loci conferring resistance to multiple virulent SCN populations had not been reported. Consistent detection of Rhg1 and Rhg4 loci with major effects and the detection of other minor effect QTL may have potentially camouflaged the epistatic interaction between these loci. Using a genetic map** strategy and testing of different SCN resistance loci combinations with multiple virulent SCN populations, we have demonstrated an epistatic interaction between rhg1-a and rhg2 that explains resistance against SCN HG type 2.5.7 (Races 1 and 5) and HG type 1.2.5.7 (Race 2) populations. SCN resistance associated with rhg2 has been previously detected in PI 89772, PI 438489B, PI 404198A, PI 437654, PI 494182, and PI 84751 (Yue et al. 2001a, b; Guo et al. 2006; Wu et al. 2009; St-Amour et al. 2020; Suzuki et al. 2020); however, none of these reports had identified its distinct role in SCN resistance through an epistatic interaction with the rhg1-a locus (Yue et al. 2001a, b; Guo et al. 2005; Wu et al. 2009; Lakhssassi et al. 2017; St-Amour et al. 2020; Suzuki et al. 2020). As such, the rhg2 gene was inadvertently reported as a minor effect QTL (Lakhssassi et al. 2017; St-Amour et al. 2020; Suzuki et al. 2020).
Emergence and expansion of virulent SCN HG type 2 populations have increased along with excessive utilization of PI 88788-type resistance (rhg1-b locus) (Niblack et al. 2008; McCarville et al. 2017; Howland et al. 2018). This breakdown of PI 88788-type resistance resulted due to the selection pressure which facilitated shifts in SCN virulence (Niblack 2005; Niblack et al. 2008; Meinhardt et al. 2021). Hence, there is an urgent necessity for the deployment of alternative sources of resistance apart from PI 88788 to manage yield losses due to SCN (McCarville et al. 2017). SCN resistance loci pyramiding has been proposed as an important strategy for SCN management (Brzostowski and Diers 2017; Yu and Diers 2017; Meinhardt et al. 2021) which requires a solid understanding of the complex interactions among SCN resistance loci from varying soybean germplasm against different SCN populations. Efforts directed at bolstering resistance to SCN populations that have overcome rhg1-b by pyramiding with G. soja QTL cqSCN-006, cqSCN-007 from PI 468916, and Chr. 10 QTL from PI 567516C SCN resistance have shown some success (Brzostowski and Diers 2017; Yu and Diers 2017; Meinhardt et al. 2021). However, recent studies have demonstrated the potential risks in the generation of more virulent SCN populations by employing SCN resistance combinations without studying the impact on SCN virulence shifts (Chen 2020; Meinhardt et al. 2021). Here, we demonstrate that pyramiding rhg1-a and rhg2 provides an effective strategy for develo** SCN resistance to cope with the rapid increase in HG type 2 and HG type 1.2 SCN field populations.
Summary
Through genetic map** approaches, we identified a major role for rhg2 in SCN resistance through a unique epistatic interaction with rhg1-a in PI 90763 that provides resistance against multiple virulent SCN populations. The stacking of rhg1-a with rhg2 provides a clear, achievable, and relatively fast solution to diversify commercially available soybean cultivars. It also offers an additional resistance rotation option for sustainable SCN management. Furthermore, the two-gene model breeding strategy recommended here is pragmatic and straightforward, which does not require testing for copy number variation of resistance loci. Thus, we propose the two-gene model as a novel breeding strategy for the next generation of SCN-resistant cultivars.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Abdel-Haleem H, Ji P, Boerma HR, Li Z (2013) An R package for SNP marker-based parent offspring tests. Plant Methods 9(1):44. https://doi.org/10.1186/1746-4811-9-44
Allen TW, Bradley CA, Sisson AJ, Byamukama E, Chilvers MI, Coker CM, Collins AA, Damicone JP, Dorrance AE, Dufault NS, Esker PD (2017) Soybean yield loss estimates due to diseases in the United States and Ontario, Canada, from 2010 to 2014. Plant Health Prog 18(1):19–27
Anand SC, Sharma SB (1995) Genetics of soybean-Heterodera glycines system. Integr Pest Manag Rev 1:65–69
Anand SC, Shumway CR (1985) Response of secondary selection on soybean cyst nematode reproduction on soybean lines. Crop Prot 4(2):231–234
Arelli AP, Wilcox JA, Myers O, Gibson PT (1997) Soybean germplasm resistant to races 1 and 2 of Heterodera glycines. Crop Sci 37(4):1367–1369
Bandara AY, Weerasooriya DK, Bradley CA, Allen TW, Esker PD (2020) Dissecting the economic impact of soybean diseases in the United States over two decades. PLoS ONE 15: e0231141
Bayless AM, Zapotocny RW, Han S, Grunwald DJ, Amundson KK, Bent AF (2019) The rhg1-a (Rhg1 low-copy) nematode resistance source harbors a Copia-family retrotransposon within the Rhg1-encoded α-SNAP gene. Plant Direct 3:1–19
Beche E, Gillman JD, Song Q, Nelson R, Beissinger T, Decker J, Shannon G, Scaboo AM (2020) Nested association map** of important agronomic traits in three interspecific soybean populations. Theor Appl Genet 133(3):1039–1054
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association map** of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brim CA (1966) A modified pedigree method of selection in soybeans. Crop Sci 6:220
Brucker E, Carlson S, Wright E, Niblack T, Diers B (2005) Rhg1 alleles from soybean PI 437654 and PI 88788 respond differentially to isolates of Heterodera glycines in the greenhouse. Theor Appl Genet 111:44–49
Brzostowski LF, Diers BW (2017) Pyramiding of alleles from multiple sources increases the resistance of soybean to highly virulent soybean cyst nematode isolates. Crop Sci 57(6):2932–2941
Caldwell BE, Brim CA, Ross JP (1960) Inheritance of resistance of soybeans to the cyst nematode Heterodera Glycines. Agron J 52(11):635–636
Chen S (2020) Dynamics of population density and virulence phenotype of the soybean cyst nematode as influenced by resistance source sequence and tillage. Plant Dis 104:2111–2122
Concibido VC, Denny R, Lange DA, Orf JH, Young N (1997) Genome map** on soybean cyst nematode resistance genes in ‘Peking’, PI 90763, and PI 88788 using DNA markers. Crop Sci 37:258–264
Concibido VC, Diers BW, Arelli PR (2004) A decade of QTL map** for cyst nematode resistance in soybean. Crop Sci 44:1121–1131
Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM, Wang J, Hughes TJ, Willis DK, Clemente TE, Diers BW (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338(6111):1206–1209
Cook DE, Bayless AM, Wang K, Guo X, Song Q, Jiang J, Bent AF (2014) Distinct copy number, coding sequence, and locus methylation patterns underlie Rhg1-mediated soybean resistance to soybean cyst nematode. Plant Physiol 165:630–647
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bulletin 19:11–15
Gardner M, Heinz R, Wang J, Mitchum MG (2017) Genetics and adaptation of soybean cyst nematode to broad-spectrum soybean resistance. G3 Genes. Genomes, Genet 7:835–841
Glover KD, Wang D, Arelli PR, Carlson SR, Cianzio SR, Diers BW (2004) Near isogenic lines confirm a soybean cyst nematode resistance gene from PI 88788 on linkage group. Crop Sci 44:936–941
Guo B, Sleper DA, Arelli PR, Shannon JG, Nguyen HT (2005) Identification of QTLs associated with resistance to soybean cyst nematode races 2, 3, and 5 in soybean PI 90763. Theor Appl Genet 111(5):965–971
Guo B, Sleper DA, Nguyen HT, Arelli PR, Shannon JG (2006) Quantitative trait loci underlying resistance to three soybean cyst nematode populations in soybean PI 404198A. Crop Sci 46(1):224–233
Howland A, Monnig N, Mathesius J, Nathan M, Mitchum MG (2018) Special report survey of Heterodera glycines population densities and virulence phenotypes during 2015–2016 in Missouri. Plant Dis 102:2407–2410
Jiao Y, Vuong TD, Liu Y, Meinhardt C, Liu Y, Joshi T, Cregan PB, Xu D, Shannon G, Nguyen HT (2015) Identification and evaluation of quantitative trait loci underlying resistance to multiple HG types of soybean cyst nematode in soybean PI 437655. Theor Appl Genet 128:15–23
Kadam S, Vuong TD, Qiu D, Meinhardt CG, Song L, Deshmukh R, Patil G, Wan J, Valliyodan B, Scaboo AM, Shannon JG (2016) Genomic-assisted phylogenetic analysis and marker development for next-generation soybean cyst nematode resistance breeding. Plant Sci J 242:342–350
Kandoth PK, Liu S, Prenger E, Ludwig A, Lakhssassi N, Heinz R, Zhou Z, Howland A, Gunter J, Eidson S, Dhroso A, LaFayette P, Tucker D, Johnson S, Anderson J, Alaswad A, Cianzio SR, Parrott WA, Korkin D, Meksem K, Mitchum MG (2017) Systematic mutagenesis of serine hydroxymethyltransferase reveals an essential role in nematode resistance. Plant Physiol 175:1370–1380
Kazi S, Shultz J, Afzal J, Hashmi R, Jasim M, Bond J, Arelli PR, Lightfoot DA (2010) Iso-lines and inbred-lines confirmed loci that underlie resistance from cultivar ‘Hartwig’ to three soybean cyst nematode populations. Theor Appl Genet 120(3):633–644
Kim M, Diers BW (2013) Fine map** of the SCN resistance QTL cqSCN-006 and cqSCN-007 from Glycine soja PI 468916. Crop Sci 53:775–785
Klepadlo M, Meinhardt CG, Vuong TD, Patil G, Bachleda N, Ye H, Robbins RT, Li Z, Shannon JG, Chen P, Meksem K (2018) Evaluation of soybean germplasm for resistance to multiple nematode species: Heterodera glycines, Meloidogyne incognita, and Rotylenchulus reniformis. Crop Sci 58(6):2511–2522
Koenning SR, Wrather JA (2010) Suppression of soybean yield potential in the continental United States by plant diseases from 2006 to 2009. Plant Health Prog 11:1. https://doi.org/10.1094/PHP-2010-1122-01-RS
Kofsky J, Zhang H, Song BH (2021) Novel resistance strategies to soybean cyst nematode (SCN) in wild soybean. Sci Rep 11:7967
Lakhssassi N, Liu S, Bekal S, Zhou Z, Colantonio V, Lambert K, Barakat A, Meksem K (2017) Characterization of the soluble NSF attachment protein gene family identifies two members involved in additive resistance to a plant pathogen. Sci Rep 7:45226
Lee TG, Kumar I, Diers BW, Hudson ME (2015) Evolution and selection of Rhg1, a copy-number variant nematode-resistance locus. Mol Ecol 24(8):1774–1791
Liu S, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, Yang C, Jamai A, El-Mellouki T, Juvale PS, Hill J (2012) A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492(7428):256
Liu S, Kandoth PK, Lakhssassi N, Kang J, Colantonio V, Heinz R, Yeckel G, Zhou Z, Bekal S, Dapprich J, Rotter B (2017) The soybean GmSNAP18 gene underlies two types of resistance to soybean cyst nematode. Nature Commun 8(1):14822
Luedders VD, Dropkin BH (1983) Effect of secondary selection on cyst nematode reproduction on soybeans. Crop Sci 23:263–264
McCarville MT, Marett CC, Mullaney MP, Gebhart GD, Tylka GL (2017) Increase in soybean cyst nematode virulence and reproduction on resistant soybean varieties in Iowa from 2001 to 2015 and the effects on soybean yields. Plant Health Prog 18:146–155
Meinhardt CG, Howland A, Ellersieck M, Scaboo AM, Diers BW, Mitchum MG (2021) Resistance gene pyramiding and rotation to combat widespread soybean cyst nematode virulence. Plant Dis 105(10):3238–3243. https://doi.org/10.1094/PDIS-12-20-2556-RE
Meksem K, Pantazopoulos P, Njiti VN, Hyten LD, Arelli PR, Lightfoot DA (2001) ‘Forrest’ resistance to the soybean cyst nematode is bigenic: saturation map** of the Rhg1 and Rhg4 loci. Theor Appl Genet 103:710–717
Mitchum MG (2016) Soybean resistance to the soybean cyst nematode Heterodera glycines: An update. Phytopathology 106:1444–1450
Myers GO, Anand SC (1991) Inheritance of resistance and genetic relationships among soybean plant introductions to races of soybean cyst nematode. Euphytica 55:197–201
Niblack TL (2005) Soybean cyst nematode management reconsidered. Plant Dis 89(10):1020–1026
Niblack TL, Arelli PR, Noel GR, Opperman CH, Orf JH, Schmitt DP, Shannon JG, Tylka GL (2002) A revised classification scheme for genetically diverse populations of Heterodera glycines. J Nemat 34(4):279
Niblack TL, Lambert KN, Tylka GL (2006) A model plant pathogen from the kingdom Animalia: Heterodera glycines, the soybean cyst nematode. Annu Rev Phytopathol 44:283–303
Niblack TL, Colgrove AL, Colgrove K, Bond JP (2008) Shift in virulence of soybean cyst nematode is associated with the use of resistance from PI 88788. Plant Health Prog 9(1):29–33
Niblack TL, Tylka GL, Arelli P, Bond J, Diers B, Donald P, Faghihi J, Gallo K, Heinz RD, Lopez-Nicora H, Von Qualen R (2009) A standard greenhouse method for assessing soybean cyst nematode resistance in soybean: SCE08 (standardized cyst evaluation 2008). Plant Health Prog 10(1):33–41
Reuscher S, Furuta T (2016) ABHgenotypeR: easy visualization of ABH genotypes (R package ABHgenotypeR). Bioscience and Biotechnology Center, Nagoya University, 464–8601, Japan.
Schmitt DP, Shannon G (1992) Differentiating soybean responses to Heterodera glycines races. Crop Sci 32(1):275–277
Song Q, Yan L, Qungley C, Fickus E, Wei H, Chen L, Dong F, Araya S, Liu J, Hyten D, Pantalone V, Nelson RL (2020) Soybean BARCSoySNP6K: An assay for soybean genetics and breeding research. Plant J 104(3):800–811
SoyBase (2021) Soy Base Database. https://www.soybase.org/sbt/search/search_results.php?category=TraitName&search_term=Reaction+to+Heterodera+glycines. Accessed 15 Mar 2021
St-Amour VTB, Mimee B, Torkamaneh D, Jean M, Belzile F, O’Donoughue LS (2020) Characterizing resistance to soybean cyst nematode in PI 494182, an early maturing soybean accession. Crop Sci 60:2053–2069. https://doi.org/10.1002/csc2.20162
Suzuki C, Taguchi-Shiobara F, Ikeda C, Iwahashi M, Matsui T, Yamashita Y, Ogura R (2020) Map** soybean rhg2 locus, which confers resistance to soybean cyst nematode race 1 in combination with rhg1 and Rhg4 derived from PI 84751. Breed Sci 70(4):474–480
Takuno S, Terauchi R, Innan H (2012) The Power of QTL Map** with RILs. PLoS ONE 7(10):e46545. https://doi.org/10.1371/journal.pone.0046545
Thomas JD, Caviness CE, Riggs RD, Hartwig EE (1975) Inheritance of reaction to race 4 of soybean cyst nematode. Crop Sci 15:208–210
Tylka GL (2016) Understanding soybean cyst nematode HG types and races. Plant Health Prog 17:149–151. https://doi.org/10.1094/PHP-PS-16-0615
Tylka GL, Marett CC (2021) Known distribution of the soybean cyst nematode, Heterodera glycines, in the United States and Canada in 2020. Plant Health Prog 22(1):72–74
Usovsky M, Ye H, Vuong TD, Patil GB, Wan J, Zhou L, Nguyen HT (2021a) Fine-map** and characterization of qSCN18, a novel QTL controlling soybean cyst nematode resistance in PI 567516C. Theor Appl Genet 134:621–631
Usovsky M, Lakhssassi N, Patil GB, Vuong TD, Piya S, Hewezi T, Robbins RT, Stupar RM, Meksem K, Nguyen HT (2021b) Dissecting nematode resistance regions in soybean revealed pleiotropic effect of soybean cyst and reniform nematode resistance genes. Plant Genome 14(2):e20083. https://doi.org/10.1002/tpg2.20083
van Ooijen JW (2004) MapQTL®5, Software for the map** of quantitative trait loci in experimental populations. Kyazma B.V., Wageningen, Netherlands
Voorrips R (2002) MapChart: Software for the graphical presentation of linkage maps and QTL. J Hered 93:77–78
Vuong TD, Sleper DA, Shannon JG, Nguyen HT (2010) Novel quantitative trait loci for broad-based resistance to soybean cyst nematode (Heterodera glycines, Ichinohe) in soybean PI 567516C. Theor Appl Genet 121:1253–1266
Wu X, Blake S, Sleper DA, Shannon JG, Cregan P, Nguyen HT (2009) QTL, additive and epistatic effects for SCN resistance in PI 437654. Theor Appl Genet 118(6):1093–1105
Xavier A, Xu S, Muir WM, Rainey KM (2015) NAM: association studies in multiple populations. Bioinformatics 31: btv448
Yu N, Diers BW (2017) Fine map** of the SCN resistance QTL cqSCN-006 and cqSCN-007 from Glycine soja PI 468916. Euphytica 213(2):54–58
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association map** in maize. Genetics 178:539–551
Yue P, Arelli PR, Sleper DA (2001a) Molecular characterization of resistance to Heterodera glycines in soybean PI 438489B. Theor Appl Genet 102(6–7):921–928
Yue P, Sleper DA, Arelli PR (2001b) Map** resistance to multiple races of Heterodera glycines in soybean PI 89772. Crop Sci 41:1589–1595. https://doi.org/10.2135/cropsci2001.4151589x
Zhou L, Song L, Lian Y, Usovsky M, Wan J, Vuong TD, Nguyen HT (2021) Genetic characterization of qSCN10 from an exotic soybean accession PI 567516C reveals a novel source conferring broad-spectrum resistance to soybean cyst nematode. Theor Appl Genet 134:859–874
Acknowledgements
The authors acknowledge funding for this research from the Missouri Soybean Merchandising Council, North Central Soybean Research Program, and United Soybean Board. We heartily thank personnel from the Northern Soybean Breeding Program at the University of Missouri for their contributions in conducting experiments on this research.
Author information
Authors and Affiliations
Contributions
PB conducted field experiment, phenoty**, and qtl map**; MU conducted qtl map** and marker analysis; CGM coordinated greenhouse phenoty**; AMS and MGM designed the experiments, acquired funding, and supervised the work; QS performed the genoty**; JDG helped with qtl map** analysis; PB, MU, and AMS wrote the paper; CGM, TJ, QS, BWD, and MGM revised the manuscript. All authors read the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Volker Hahn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Basnet, P., Meinhardt, C.G., Usovsky, M. et al. Epistatic interaction between Rhg1-a and Rhg2 in PI 90763 confers resistance to virulent soybean cyst nematode populations. Theor Appl Genet 135, 2025–2039 (2022). https://doi.org/10.1007/s00122-022-04091-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-022-04091-2