
Abstract
Lipid metabolic reprogramming is an emerging hallmark of cancer. In order to sustain uncontrolled proliferation and survive in unfavorable environments that lack oxygen and nutrients, tumor cells undergo metabolic transformations to exploit various ways of acquiring lipid and increasing lipid oxidation. In addition, stromal cells and immune cells in the tumor microenvironment also undergo lipid metabolic reprogramming, which further affects tumor functional phenotypes and immune responses. Given that lipid metabolism plays a critical role in supporting cancer progression and remodeling the tumor microenvironment, targeting the lipid metabolism pathway could provide a novel approach to cancer treatment. This review seeks to: (1) clarify the overall landscape and mechanisms of lipid metabolic reprogramming in cancer, (2) summarize the lipid metabolic landscapes within stromal cells and immune cells in the tumor microenvironment, and clarify their roles in tumor progression, and (3) summarize potential therapeutic targets for lipid metabolism, and highlight the potential for combining such approaches with other anti-tumor therapies to provide new therapeutic opportunities for cancer patients.
Similar content being viewed by others

Introduction
Metabolic reprogramming has emerged as a critical feature of cancer. To adapt to the hypoxic and nutrient-poor microenvironment, in addition to increasing glucose uptake and aerobic glycolysis, tumor cells also undergo lipid metabolism reprogramming to enhance their biological behaviors [1]. This is characterized by increased lipid uptake, lipid synthesis, fatty acid oxidation (FAO), and lipid storage. Mounting evidence demonstrated that lipids play a critical role in cancer progression by serving as energy sources, membrane structures, signaling molecules (including bioactive lipids like S1P, PGE2, and LPA), and even causing epigenetic modifications through fatty acylation of key molecules [2, 3]. Mechanically, alterations in lipid metabolic phenotype in tumor cells are directly driven by continuous oncogenic events and extracellular tumor microenvironment (TME) factors such as hypoxia, acidosis, and nutritional alterations [4, 5].
In addition to supporting tumor development, lipid metabolic reprogramming also modifies the TME by influencing the recruitment, activation, and function of immune cells and stromal cells. Tumor cells and cells in TME interact with each other and form a reciprocal entity [6]. On one hand, tumor cells can actively modify the TME by secreting signaling molecules and metabolites, which affect the functions of cancer-associated fibroblasts (CAFs) and immune cells in TME [6]. On the other hand, lipid metabolic reprogramming, an adaptive change in cells within the TME, manifests as increased lipid uptake and accumulation, or FAO, driving the TME toward an immunosuppressive phenotype supporting tumor progression [7]. For example, upregulated lipid uptake and FAO increase lipid metabolic levels in regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), facilitating their immunosuppressive function [8,9,10]. Moreover, upregulation of CD36 in CD8+ T cells leads to excessive lipid accumulation, which impairs secretion of anti-tumor factors such as IFN-γ and TNF-α, ultimately suppressing their anti-tumor efficacy [11, 12]. Similarly, upregulation of CD36 in natural killer (NK) cells also impairs their tumor-killing activity through intracellular lipid accumulation. Studies have suggested that blocking lipid uptake via inhibition of CD36 on cytotoxic CD8+ T cells or Tregs enhances anti-tumor immune responses [8, 11].
Given the critical role of lipids in cancer progression, targeting to lipid metabolism-related pathways offers new therapeutic opportunities for cancer. A large body of evidence shows that inhibitors targeting lipid uptake, lipogenesis, and FAO in tumor cells have shown significant therapeutic effects in various cancers [13,14,15]. Besides, modulating lipid metabolism in stromal cells and immune cells also provides a new choice for anti-tumor therapy. Moreover, it can be combined with chemotherapy and immunotherapy, providing a new comprehensive strategy for optimizing cancer treatment. This review aims to clarify the lipid metabolic landscape in tumor cells and TME cells, and summarize potential targets to offer clues for further research and clinical applications of targeting lipid metabolism in cancer.
Landscape and mechanisms of lipid metabolic reprogramming in cancer
Lipid metabolic reprogramming in cancer
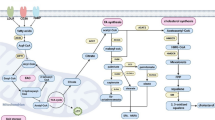
Most lipid molecules in the human diet are triacylglycerols (TAGs) and cholesterol. After absorption, TAGs can be hydrolyzed into glycerol and fatty acids (FAs). Glycerol is then converted into glycerol-3-phosphate (G-3-P), which enters glycolysis. FAs can either be stored as the primary component of membrane synthesis or converted to acyl-CoA for β-oxidation to provide energy. In tumors, several steps of lipid metabolism show universal enhancement to maintain their biological progressions. This includes increased lipid uptake, synthesis, storage, and FAO. To delve deeper into these lipid metabolic alterations, the following section provides an in-depth analysis (Fig. 1).

Lipid metabolic reprogramming in cancer. Tumor cells enhance lipid metabolism by increasing exogenous lipid uptake and lipid synthesis, leading to increased intracellular lipid content. Upregulation of lipid transport proteins such as CD36, FATPs, FABPs, and LDLR increases lipid uptake. These upregulations increase intracellular SUFA, PUFA, and cholesterol levels. Meanwhile, endogenous lipid synthesis originates from citrate in the TCA cycle, as well as intracellular glutamine, lactate and acetate, leading to the synthesis of SFA and cholesterol. The process is catalyzed by key enzymes such as FASN and SCD. FAs in tumor cells are catalyzed by ACSL to form acyl-CoA, which is involved in the subsequent synthesis of intracellular phospholipids with bioactive lipids and FAO. Acyl-CoA facilitates the translocation of enzymes into mitochondria through CPT1, the key enzyme of FAO, participating in the production of acetyl-CoA, providing energy for the biological behavior of tumor cells. Excess lipids in tumor cells are stored in LD as CE and TAG. This storage significantly prevents LPO and attenuates its risk of mediating tumor cell death
Lipid uptake in cancer
The increase in intracellular lipid content can be achieved through two pathways: endogenous and exogenous pathways. Endogenous lipids are primarily produced through de novo lipogenesis (DNL), which utilizes acetyl-CoA as a substrate. Exogenous lipids require the involvement of transport molecules, including CD36, fatty acid transport protein family (FATPs/SLC27), and fatty acid-binding proteins (FABPs) [4]. Notably, recent studies have established a link between the overexpression of these transport molecules and the poor prognosis across various cancers. For instance, CD36 overexpression is associated with a poor prognosis in breast, ovarian, gastric, colorectal, and prostate cancer [15]. Moreover, FABPs have been found to contribute to the promotion of cervical cancer metastasis by increasing intracellular Fas [16], while knockdown of FABPs suppressed tumor progression in vivo by inhibiting lipid uptake in glioblastoma [17]. FATP members have been implicated in cancer initiation and progression in melanoma and breast cancer in multiple studies [18, 19].
Lipid synthesis in cancer
Although the exogenous lipid sources increase, cancer cells also activate DNL to respond to their high metabolic demands[2]. This pathway begins with acetyl-CoA, which can be mainly generated from citrate, a substrate in the TCA cycle during nutrient catabolism, via ATP-citrate lyase (ACLY). Besides, acetate conversion via acetyl-CoA synthetase (ACSS) is another pathway to produce acetyl-CoA for DNL. Acetyl-CoA is activated by acetyl-CoA carboxylases (ACCs) to form malonyl-CoA, which is further catalyzed by fatty acid synthase (FASN) to form saturated fatty acids (SFA), palmitate (C16:0). The resulting palmitate can be elongated by elongation of very-long-chain fatty acids gene family (ELOVLs) and desaturated by stearoyl-CoA desaturases (SCDs) or fatty acid desaturases (FADSs) to synthesize monounsaturated fatty acid (MUFA), such as oleic acid (OA) (C18:1) and palmitoleic acid (C16:1). Moreover, desaturation caused by ELOVLs and FADSs converts ingested polyunsaturated fatty acids (PUFAs) like linoleic acid (LA) (C18:2) and alpha-linolenic acid (ALA) (C18:3) into other PUFAs like arachidonic acid (AA) (C20:4) and adrenic acid (AdA) (C22:4) [20, 21]. Interestingly, overexpression or increased activity of ACLY has been correlated with the progression of various cancers [22]. ACSS is transcriptionally upregulated by SREBP, highly expressed in tumor cells, and plays a role in maintaining cancer cell growth under nutrient deficiency by catalyzing acetate [23]. FASN is commonly overexpressed in many epithelial and precancerous lesions and is associated with a high risk of cancer recurrence and mortality [24]. Inhibition of FASN can suppress breast cancer growth in the brain, highlighting its potential as a therapeutic target for metastasis in breast cancer [25]. SCD1 facilitates the formation of MUFAs, including OA, and its increased expression has been shown to promote the progression of cancers [26, 27].
In oncogenic processes, tumor cells utilize other metabolic substances in the microenvironment, such as glutamine and lactate, as sources of lipid synthesis. Glutamine dependence has been considered a metabolic hallmark of cancer cells. A growing body of evidence has shown that glutamine uptake and synthesis is upregulated in various cancers [28]. Cellular glutamine undergoes a transformation into α-ketoglutarate through the activation of glutaminase (GLS) and glutamate dehydrogenase (GLUD), ensuring the replenishment of vital metabolic intermediates within the TCA cycle. Subsequently, α- ketoglutarate is carboxylated by isocitrate dehydrogenase (IDH) to generate citrate [29]. In addition to glutamine, lactate is also an important source of TCA cycle intermediates and acetyl-CoA [30]. A recent study identified that lactate in the TME can reprogram lipid metabolism by increasing the expression of the genes involved, promoting tumor progression [31]. Notably, lactate promotes glutamine uptake and catabolism in oxidative cancer cells [32]. Therefore, utilizing glutamine and lactate to produce acetyl-CoA as a source of lipid synthesis is one of the important indirect ways for tumor cells to regulate lipid metabolism.
The synthesis of triacylglycerol (TAG) from long-chain fatty acids (LCFAs) derived from lipid intake and DNL involves a series of enzymatic reactions. Specifically, glycerol-3-phosphate acyltransferase (GPAT) catalyzes the combination of LCFAs with G-3-P to generate lysophosphatidic acid (LPA), which is a crucial intermediate in TAG synthesis. LPA is then converted to diacylglycerol (DAG) and subsequently to TAG via diacylglycerol acyltransferase (DGAT) [33]. Notably, DAG is also involved in compound lipid synthesis, such as cholesterol and phospholipids, which play critical roles in supporting key oncogenic functions and cancer hallmarks, and in regulating intercellular communication and immune responses [34].
Cholesterol, like other lipids, relies on acetyl-CoA for intracellular synthesis. Activating key enzymes in the mevalonate (MVA) synthesis pathway, such as HMG-CoA reductase (HMGCR), enables cholesterol biosynthesis. MVA is further modified to generate a variety of cholesterol for important biological processes such as membrane biosynthesis. Excess cholesterol is eliminated from the cell through ATP-binding cassette transporter A1 (ABCA1) [35]. In addition, low-density lipoproteins (LDLs) are taken up through membrane receptors (LDLRs), and high LDLRs levels promote LDL cholesterol-mediated breast cancer growth [36]. Reprogramming of cholesterol metabolism in tumors is mainly characterized by increased levels of intracellular cholesterol synthesis and abnormal metabolite accumulation [35]. This upregulation of cholesterol metabolism in both tumor cells and TME can promote oncogenic processes, such as tumor initiation, migration, and angiogenesis [37, 38].
Phospholipid (PL) synthesis, using DAG as a precursor, is enhanced in cancer, which regulates biological behaviors such as metastasis and drug resistance by modulating changes in membrane lipid composition and producing bioactive lipid second messengers [39]. Phosphatidylcholine (PC) is the predominant phospholipid in most cellular membranes. An increase in PC synthesis, along with elevated levels of choline cycle metabolites such as choline, phosphocholine (PCho), and glycerophosphocholine (GPC), has emerged as a significant hallmark of malignant transformation in tumors [40]. PC metabolic enzyme, choline kinase (ChoK), has been observed to be activated in various cancers [41]. Studies both in vitro and in vivo have shown that overexpression of ChoKα contributes to tumor progression, metastasis, and aggressiveness [42, 43].
In addition, PL catabolism is mediated by phospholipases (PLA2, C, and D), which can be recycled for PL biosynthesis and modulate various lipid-mediated signaling pathways promoting tumorigenesis. PL can also be hydrolyzed by PLC and PLD, producing DAG and phosphatidic acid (PA). This sustains the activity of key oncogenic signaling pathways involving PKC and mTOR [44]. Importantly, a significant portion of PL is hydrolyzed by PLA2, leading to the production of lysophosphatidylcholine (LPC) and AA. Subsequently, under catalysis of a series of enzymes, various lipid-derived mediators, including LPA, PGs, LTs, and S1P, are generated within tumor cells. LPC is catalyzed by lysophosphatidylcholine acyltransferases (LPCATs) to be reconverted into PC. Under the action of autotaxin (ATX), LPC is converted to LPA [45]. Similarly, AA participates in lipid mediator biosynthesis, producing prostaglandins (PGs) through cyclooxygenase (COX) and leukotrienes (LTs) through lipoxygenase (LOX). And sphingosine-1-phosphate (S1P) is also derived from sphingomyelin. These lipid-derived mediators are released extracellularly and act as crucial signaling molecules that mediate the crosstalk between the tumor and the TME for cancer progression [46].
Lipid storage in cancer
Increased uptake and endogenous synthesis of lipids in tumor cells lead to an increase in the cellular lipid pool. Acyl-CoA cholesterol acyltransferase (ACAT) converts free cholesterol to cholesteryl ester (CE) within the endoplasmic reticulum (ER) membrane, while excess intracellular FAs are ultimately converted into TAG by DGAT. These lipids are then stored as CE and TAG within lipid droplets (LD) in cells, reducing cell damage caused by peroxidation of free lipids within the cell [47, 48]. Lipids stored in LD can provide ATP response to metabolic stress by undergoing β-oxidation to produce acetyl-CoA. LDs serve as a critical reservoir of unsaturated FAs that cancer cells can use to maintain the function of cell membranes and organelles, particularly when there is an increased demand for lipids, such as during rapid oncogene-driven cell growth or a hypoxic environment [48]. Additionally, another key function of LDs is to protect cancer cells under ER stress and oxidative stress [49]. DGAT1, a key protein in lipid accumulation, promoting LDs formation and protecting cancer against lipid peroxidation, has been found to play indispensable oncogenic roles in melanoma and glioblastoma [50, 51].
Lipolysis in cancer
Degradation of TAG in LD can be initiated by adipose triglyceride lipase (ATGL), hydrolyzing TAG to produce DAG. DAG is then hydrolyzed by hormone-sensitive lipase (HSL) and monoacylglycerol lipase (MAL) to release FFA. Given that lipid synthesis and LD accumulation are common metabolic characteristics of cancers, ATGL, the key enzyme for LD mobilization, is generally downregulated in most cancer cells [52]. Recent research suggests that enhanced ATGL expression exerts an anti-tumor effect in triple-negative breast cancer cells [53]. In hypoxic cancer cells, LD is significantly accumulated, which is resulted by the activation of hypoxia-inducible gene 2 (HIG2) to inhibit ATGL-mediated lipolysis [54, 55]. However, ATGL upregulation in tumor exhibits pro-tumor effects in some adipose-infiltrated cancers, including colorectal and breast cancer [56, 57]. In coculture systems, breast cancer cells exhibit increased proliferation and migration after acquiring FAs from adipocytes, which is dependent on the lipolysis induced by ATGL in both adipocytes and cancer cells [57]. Besides, ATGL also play a crucial role in the development of colon cancer driven by obesity [58]. These studies indicate that lipolysis acts as a double-edged sword in cancer progression, and the underlying mechanisms require further elucidation.
Lipid oxidation in cancer
As FAs uptake and storage increase, FAs catabolism in cancer cells is often enhanced. The survival and metastasis of cancer cells also rely on the uptake and consumption of FAs. FAO serves as an energy source for tumor cells under nutrient-deficient conditions. Carnitine palmitoyl transferase 1 (CPT1), the rate-limiting enzyme involved in mitochondrial FAO of LCFAs, mediates the entry of FAs into mitochondria. Once FAs enter the mitochondrial matrix, they are oxidized to generate acetyl-CoA, which enters the TCA cycle to produce ATP. The long-chain acyl-CoA synthase (ACSL) enzyme family plays an important role in FAO and lipids biosynthesis, facilitating the production of fatty acyl-CoA [59]. Tumors exhibit high FAO activity by upregulating CPT1A expression. Moreover, upregulation of CPT1A expression can promote EMT and stemness, leading to the invasive and metastatic capabilities of cancer cells [60, 61].
Lipid peroxidation (LPO) is prone to occur in PUFA-phospholipids (PUFA-PLs), resulting in the accumulation of lipid peroxidation within cells, which is caused by an imbalance in the ratio of intracellular PUFA to MUFA. This phenomenon is commonly associated with an increase in PUFA due to LD synthesis inhibition or a decrease in MUFA due to downregulation of enzyme activity involved in MUFA synthesis. LPO is significant in mediating ferroptosis and is often inhibited in progressing tumors [62, 63]. LDs, essential mediators of free unsaturated FA (especially PUFA) storage, regulate LPO and susceptibility to ferroptosis [64]. Upregulation of DGAT promotes LD synthesis in glioblastoma and gastric cancer cells. Inhibiting the formation of LDs by silencing DGATs can induce LPO and ferroptosis, thereby inhibiting cancer cell metastasis [50, 65]. Maintaining MUFA-phospholipid (MUFA-PL) levels in the cell membrane is critical for tumor cells to avoid ferroptosis. ASCL3, which is upregulated in various cancers, mainly catalyzes MUFA generation to form fatty acyl-CoA, promoting the synthesis of MUFA-PLs [59]. Furthermore, ACSL4 promotes the increase of membrane PUFA-PL levels by acting on PUFA, an essential therapeutic approach for tumor by increasing LPO and inducing ferroptosis [66]. Inhibition of the key enzyme SCD1 in gastrointestinal cancers reduces MUFA production, inducing ferroptosis and exerting anti-tumor effects [67, 68]. As research progresses, inducing the accumulation of lipid peroxides and promoting ferroptosis have become potential targets for anti-tumor therapy through lipid metabolism.
Oncogenic cues affecting tumor lipid metabolism
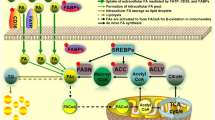
Activation of oncogenes and loss-function of tumor suppressor genes are the main causes of tumorigenesis. They also play an important role in reprogramming tumor metabolism by regulating lipid metabolic enzyme expression [4]. Sterol regulatory element-binding proteins (SREBPs) act as key upstream regulators of lipid metabolism. SREBP is a transcription factor that promotes DNL by upregulating key enzymes such as ACLY, FASN, and SCD, which are closely linked to tumor proliferation, apoptosis, and invasion [69]. Moreover, SREBP maintains intracellular cholesterol levels by inducing LDL receptor-mediated cholesterol uptake and inhibiting ABCA1-mediated cholesterol export in a mTORC1-dependent manner [70]. Downstream lipid reprogramming events are induced by SREBPs and mutations in oncogenes such as PI3K and MYC, as well as tumor suppressor genes such as p53 and PTEN (Fig. 2a).

Lipid metabolic alterations induced by oncogenic events and TME cues. Alterations in lipid metabolism in tumor cells are driven by oncogenic events and tumor environmental cues, such as hypoxia and nutritional deficiency conditions. a Activating mutations in oncogenes (such as MYC) and inactivation of tumor suppressor genes (such as p53 and PTEN) often occur in various tumor cells. Alterations in these genes subsequently upregulate the expression of key enzymes involved in lipid metabolism by regulating the PI3K-AKT-mTOR signaling pathway to activate transcriptional activity of SREBPs, or acting directly on key enzymes. As a consequence, lipid metabolism in tumor cells is enhanced. b TME encompasses various factors such as hypoxia, acidosis, and nutrient deficiency that promote tumorigenesis and cancer progression through reprogramming lipid metabolism in tumors. Lipid uptake, synthesis, and intracellular lipid accumulation are significantly upregulated in TME by activating key signaling pathways and enzymes
PI3K mutation
The dysregulation of the PI3K-AKT signaling pathway is a frequent occurrence in cancer and leads to metabolic reprogramming, where SREBPs play a crucial role as downstream regulatory targets. This pathway can be activated by various upstream signaling events, such as receptor tyrosine kinase (RTK) signaling or oncogenic mutations in PIK3CA [71]. By increasing lipid synthesis, this pathway promotes the occurrence and progression of liver cancer [72, 73]. The tumor suppressor PTEN is a critical negative regulator of the PI3K/AKT pathway, and its mutation can also activate this pathway [74]. Research has demonstrated that the loss of PTEN and the activation of PI3K/AKT lead to LD synthesis, contributing to prostate cancer progression [75]. In xenograft mice models of PIK3CA mutant breast cancer and PTEN-deficient prostate cancer, the excessive activation of the PI3K-AKT-mTOR signaling protects cancer cells from LPO and ferroptosis by SREBP1/SCD1-mediated MUFA synthesis [69]. Similarly, recent studies have shown that aspirin inhibits mTOR/SREBP-1/SCD1-mediated MUFA production and induces ferroptosis in PIK3CA mutant colorectal cancer [76]. In summary, the PI3K-AKT signaling may prove to be a potential therapeutic strategy for treating cancer as a metabolic disease.
P53 mutation
TP53, which encodes the tumor suppressor p53, is a commonly mutated oncogene in human cancers. p53 can bind directly to the promoter region of SREBP-1 and transcriptionally inhibit its expression, affecting downstream expression of key enzymes (ACLY, FASN) involved in lipogenesis [77]. Additionally, p53 suppresses the pentose phosphate pathway (PPP), which decreases NADPH production required for lipid synthesis [78, 79]. In breast cancer, p53 mutants have been shown to promote cancer progression by increasing cholesterol synthesis through enzymes involved in upregulation of the mevalonate pathway, highlighting the potential to target this pathway for p53-mutated tumors [80]. Moreover, the expression of genes key to FA synthesis (FASN, ELOVL6, and SCD1) is increased in p53-mutated tumors, while p53 transcription induces CPT1, which increases FAO and reduces intracellular lipid accumulation [81].
MYC mutation
MYC, a commonly activated oncogene in tumors, is known to promote transcriptional activation of genes involved in cell cycle, cell growth, and metabolism [82]. In addition to activating SREBP1, MYC directly regulates the expression of key enzymes involved in FA synthesis, such as ACLY, ACC, FASN, and SCD1, which have been shown to drive tumorigenesis in hepatocellular carcinoma (HCC) [83,84,85]. MYC also cooperates with SREBP2 to upregulate HMGCR for cholesterol metabolic reprogramming, contributing to the malignant phenotypes of tumor cells [86, 87]. Moreover, MYC-driven cancer cells exhibit enhanced glutamine utilization, with increased expression of key glutamine-metabolizing enzymes, including GLS1 and GLUD1, as well as the transporter protein SLC1A5 [88]. This augmented glutamine catabolism results in mitochondrial metabolic reprogramming to accommodate the replenishment requirements of the TCA cycle, supplying substrates for DNL, thereby sustaining cell vitality and growth [89]. What’s more, breast cancer cells with MYC overexpression show increased dependence on FAO for bioenergetics. Inhibiting FAO markedly diminishes the energy metabolism of these cells, suggesting that targeting FAO could be a potential therapeutic strategy for breast cancer [90, 91].
Microenvironment factors affecting tumor lipid metabolism
Metabolic reprogramming of cancer cells is the result of a multifactorial process. Along with the activation of oncogenic signals caused by mutations in tumor cells, TME also plays a crucial role [92]. The TME encompasses various factors such as hypoxia, acidosis, and nutritional deficiencies, which promote tumor initiation and cancer progression by altering lipid metabolism in tumor cells (Fig. 2b).
Hypoxia
The rapid proliferation of solid tumors consumes a large amount of oxygen, leading to hypoxia as a typical feature of almost all TME [93]. The resulting hypoxia inhibits the pyruvate metabolic pathway of glucose, resulting in decreased citrate content in the TCA cycle. As citrate, the primary substrate for DNL, decreases, cancer cells turn to alternative carbon sources such as glutamate or acetate to produce acetyl-CoA for FA synthesis [2, 94]. Hypoxic tumor cells utilize glutamine and synthesize citrate under IDH1 catalysis, a process that relies on the expression of hypoxia-inducible factor (HIF1) [95, 96].
In addition to alterations in glucose metabolism, lipid metabolism also undergoes changes in hypoxic tumor cells. FA catabolism is dependent on oxygen, and tumor cells often inhibit FAO through various pathways. Hypoxia-activated HIF-1α and HIF-2α downregulate CPT-1 expression, which prevents FAs from entering the mitochondria for β-oxidation [97, 98]. As a result, FAs are redirected to LD storage, leading to increased lipid accumulation [99]. Additionally, HIF-1α upregulates FABPs expression in hypoxia, promoting FA uptake and lipid storage by regulating the expression of key enzymes involved in TAG synthesis [17, 100]. In clear cell renal cell carcinoma (ccRCC), hypoxia increases intracellular LD synthesis in a HIF-2α-dependent manner, which plays a crucial role in sustaining ER homeostasis and aggressive tumor behaviors [101]. Furthermore, HIF-1 upregulates the expression of low-density lipoprotein receptor-related protein 1 (LRP1) to promote lipoprotein endocytosis and the lipids storage in LDs, providing energy for cells during hypoxia [102, 103].
In summary, hypoxia induces genes involved in FA uptake, synthesis, and storage, leading to an overall increase in intracellular lipid content. The inhibition of key enzymes involved in FAO also supports lipid accumulation under hypoxia, and the accumulation of lipids in LDs helps sustain malignant behaviors of tumors [104].
Acidosis
TME is characterized by hypoxia and acidosis, both of which contribute to the metabolic reprogramming of tumor cells. Hypoxia results in lactate accumulation and H+ build-up, which alter the metabolic pathways of cancer cells and promote tumor metastasis [105]. Lactate activates the expression of the SLC1A5 and GLS1 to promote glutamine transport and catabolism, which provides substrates for the TCA cycle [32]. In the absence of acetyl-CoA produced by glycolysis in an acidic microenvironment, tumors rely heavily on FAO to generate energy by converting LCFAs into acetyl-CoA and producing NADH and FADH2 [106]. Interestingly, in tumor cells, an acidic environment upregulates CD36 and DGAT expression, promoting exogenous lipid uptake and the formation of LDs, thereby promoting metastasis through a TGF-β2-dependent mechanism [107]. Acidosis also stimulates fatty acid synthesis (FAS) and promotes hepatocarcinogenesis through activating the PI3K/AKT pathway, upregulating SCD1, and promoting its binding to peroxisome proliferator activated receptor-α (PPARα) [108]. Additionally, an acidic TME triggers SREBP2 activation in tumors, leading to upregulation of the downstream key enzyme ACSS2, which provides suitable growth conditions for cancer cells in the acidic environment. These findings suggest that an acidic TME and SREBP2 activation is associated with reduced overall survival of cancer patients [109].
Nutrient alteration
The rapid proliferation of tumors requires continuous acquisition of nutrients from the microenvironment, resulting in a nutrient-deficient TME[105, 110]. In obese phenotypes, changes in metabolic spectra within tumor tissues result in the accumulation of lipids in adipocytes within the TME [111, 112]. Consequently, tumor cells must alter their metabolic patterns to enhance the utilization of lipids for energy supply to sustain their biological behaviors. In fat-rich TME, cancer cells can produce cytokines that induce ATGL-dependent lipolysis in the adipocytes surrounding the tumor, resulting in the release of FFAs [113]. These FFAs are then taken up by cancer cells via CD36 and FABP3/4 to form LDs, which act as energy sources for malignant cells. Moreover, lipid transfer from adipocytes to cancer cells can also be facilitated by extracellular vesicles (EVs) [114, 115]. Adipocytes in the TME have been shown to act as metabolic regulators that promote the growth and survival of colon cancer cells [116]. When co-cultured with breast cancer cells, the consumption of TAGs within adipocytes is increased, which in turn transfers adipocyte-derived FFAs to breast cancer cells, increasing CPT1A levels and driving FA metabolism in cancer cells [117].
Dietary factors affecting tumor lipid metabolism
Dietary interventions can change metabolite levels in the TME, which may then affect cancer cell metabolism to alter tumor growth [118]. Obesity and excessive high-fat diets (HFDs) are associated with increased overall and cancer-specific mortality, especially among patients with breast, colon and uterine cancer [119]. Excessive adipose expansion during obesity causes adipose dysfunction and inflammation to increase systemic levels of proinflammatory factors. Cancer-associated adipocytes can enter the TME to enhance pro-tumor effects [120]. In obese mouse models, HFDs increase the number of LGR5+ intestinal stem cells and activate the lipid metabolism transcription factor PPARγ for their tumorigenic potential [121]. Importantly, fat-mediated inflammatory signaling and the immunosuppressive microenvironment in tumors are the main causes of tumor metastasis [122]. For instance, HFD-induced obesity leads to CD8+ T cell exhaustion by reducing the production of granzymes and cytokines (IFN-γ and TNF-α), ultimately accelerating tumor growth in mouse models [123, 124]. Moreover, HFDs can induce lipid accumulation in prostate tumors in a SPREP-dependent manner, facilitating metastasis in mouse models [125]. Similarly, chronic HFDs alter biological behaviors, including angiogenesis and proliferation, in breast cancer [126].
Notably, different types of dietary lipids exhibit heterogeneity in driving the biological behavior of tumors. A study demonstrates that dietary palmitic acid, in contrast to OA or LA, enhances metastasis in oral carcinomas and melanoma in mouse models [127]. SFA intake is associated with an enhanced MYC signaling and poorer outcome in prostate cancer patients [128]. OA can also favor survival and chemotherapy resistance in gastric cancer [129]. Mechanistically, HFDs raise systemic FA levels, including SFAs and unsaturated FAs, which further enhances FAO, producing enough energy to facilitate tumor progression. In addition, two essential FAs: ALA (ω-3 PUFA) and LA (ω-6 PUFA), play proinflammatory and anti-inflammatory roles in tumors, respectively. In patients with metastatic colorectal cancer, primary tumors exhibit significantly elevated levels of ω-6 PUFAs and reduced levels of ω-3 PUFAs compared to those in patients with non-metastatic cancer [130]. Dietary LA stimulates invasion and peritoneal metastasis of gastric cancer through COX-1-catalyzed lipid metabolism [131]. Consequently, alterations in dietary lipid consumption, adopting a low-fat eating pattern or increasing ω-3 PUFAs intake, may represent a selective anti-tumor therapy.
Landscape and mechanisms of lipid metabolic reprogramming in tme
As cancer progresses, the TME also undergoes lipid metabolic reprogramming. It is worth noting that tumor cells play a significant role in modifying TME (e.g., acidosis, lipid accumulation) by producing metabolites and lipid-related signaling molecules. This in turn influences metabolic patterns and immune phenotype of TME cells, resulting in immune microenvironment remodeling [65]. For example, the secretion of lipids by CAFs in the TME promotes tumor progression by directly supplying energy sources to tumor cells. What’s more, lipid metabolic reprogramming in CAFs also influences its own cytokine secretion function, which subsequently modulates the immune responses and promotes the formation of an immunosuppressive microenvironment. In addition, alterations in lipid metabolism patterns in immune cells also favor the construction of an immunosuppressive microenvironment, supporting tumor immune escape. Therefore, tumor progression is the result of a co-evolutionary process between tumor and TME. Moving forward, this section will focus on lipid metabolic alterations in TME cells and their interactions with tumor cells (Fig. 3).

Lipid metabolism landscape in TME. Tumor cells have the ability to educate TME cells into a pro-tumor phenotype by secreting metabolites and signaling molecules, such as cytokines and bioactive lipids, to TME, which further facilitates cancer progression. For instance, CAFs activated by tumor cells lead to increased levels of lipid synthesis, which serve as an essential energy source for tumor cells and induce the formation of an immunosuppressive microenvironment through secreting EVs and signaling molecules. Besides, tumor-infiltrating CD8+ T cells and NK cells also exhibited increased lipid uptake and FAO. Similarly, lipid accumulation in TAMs also regulates their polarization and functional phenotypes. Increased lipid uptake and FAO in immunosuppressive cells, such as Tregs and MDSCs, further enhanced their pro-tumor effects. Taken together, lipid metabolic reprogramming facilitates crosstalk between tumor cells and TME cells, fueling tumor cells and changing functional phenotypes of TME cells
Lipid metabolic reprogramming in CAFs
Cancer-associated fibroblasts (CAFs), an important stromal cell type in the TME, are activated by TGF-β and LPA signaling in TME [132, 133]. They synthesize and secrete lipids and bioactive lipid signaling molecules, playing an essential role in tumor metabolic alteration, proliferation, invasion and immune responses [134]. To adapt to the TME, CAFs undergo lipid metabolic reprogramming characterized by an increase in lipid synthesis, storage, and secretion by upregulating key enzymes like FASN and SCD [135, 136]. This constructs a microenvironment with lipid accumulation, leading to metabolic reprogramming and biological behavior enhances in tumors [115, 137].
In the hypoxic and nutrient-poor microenvironment, CAFs upregulate SCD1 expression via HIF-1α, increasing the abundance of lipids such as OA in CAFs, resulting in promoting lung cancer growth [136, 138]. Therefore, targeting SCD1 in CAFs could be a promising therapeutic strategy. Recent metabolomics studies have shown that CAF-derived lipids enhance lipid uptake in tumor cells, promoting peritoneal metastasis of colorectal cancer [139]. This phenomenon also depends on the upregulation of CPT1 in CAFs, which enhances FAO and shapes the TME for colorectal cancer metastasis [140].
In addition, CAFs shape lipid metabolism and biological behavior of tumor cells by secreting biologically active lipid molecules and EVs containing small molecules and lipid metabolites [20, 141, 142]. Overexpressed biologically active lipids, such as LPC, in CAFs can be released into the TME and absorbed by tumor cells to promote tumor proliferation and migration through intracellular lipid metabolic reprogramming [135, 143]. Mechanistically, LPC is hydrolyzed by LPA in cancer cells, activating the AKT signaling pathway [144]. PGE2 and S1P in CAFs also play vital roles in tumor progression in breast cancer and neuroblastoma [145,146,147]. What’s more, a high concentration of proinflammatory cytokines secreted by CAFs supernatant can induce upregulation of cholesterol metabolism in prostate cancer cells, promoting androgen receptor therapy resistance [148]. miR-522, secreted from CAF-derived EVs, inhibits ferroptosis in cancer by targeting arachidonate lipoxygenase 15 (ALOX15) and blocking LPO [149]. In breast cancer, CAF enhances their exogenous lipid uptake capacity by inducing upregulation of FATP1 [150, 151]. Similarly, CAFs promotes colon cancer cells to absorb lipids secreted from CAFs through CD36, thereby promoting cancer cell migration [135]. Lactate secreted by CAF induces lipid metabolic reprogramming in prostate cancer, leading to the LD formation and mobilization, concurrently enhancing tumor invasiveness [31]. These evidences suggest that the signaling molecules and metabolites secreted by CAFs play an essential role in tumor progression and may become promising therapeutic targets in future [152].
Overall, the crosstalk between CAFs and cancer cells is mediated by lipid metabolic reprogramming that contributes to cancer progression, metastasis, and therapeutic resistance. Moreover, recent studies have shown that lipid metabolic reprogramming of CAFs also plays an important role in remodeling the tumor immune microenvironment. For example, CD36+ CAFs recruit MDSCs through upregulating MIF expression, thus promoting immune escape in HCC. Inhibitors targeting to CD36 can restore the anti-tumor immune response in HCC and synergistically enhance the anti-tumor effect of anti-PD-1 therapy [153].
Lipid metabolic reprogramming in immune cells
Lipids are critical metabolites that support the biological activities of immune cells. Under normal conditions, immune cells with anti-tumor activity, such as effector CD8+ T cells, NK cells, and M1 macrophages, depend on glycolysis for their maturation and function, whereas immune-regulatory cells such as Tregs, M2 macrophages, and MDSCs rely on FAO to exert their tumor immune suppression effects [115]. When tumors occur, lipid metabolism of tumor cells and stromal cells, such as adipocytes and CAFs, contributes significantly to the establishment of a TME characterized by low glucose and high lipid accumulation [115, 137, 154]. In such a TME, immune cells display increased immunosuppressive effects by regulating their lipid metabolism patterns, and subsequently promoting tumor progression [65]. For instance, lipid accumulation and enhanced FAO in TAMs contributed to its polarization to the M2 phenotype, which blocked anti-tumor T cell responses and supported the immunosuppressive function of T cells [155]. Increased uptake of oxidized lipids and enhanced lipid peroxidation in CD8+ tumor-infiltrating lymphocytes (TILs) lead to their immune dysfunction, whereas lipid peroxidation resolution restored the functionalities of CD8+ TILs in vivo [11] (Table 1).
CD8 + T cells
CD8+ TILs are an important component of anti-tumor immune cells, but their cytotoxic effects change to an exhausted state as cancer progresses [156,157,158]. In response to the limited nutrients and glucose in the TME, CD8+ TILs experience a significant reduction in glycolytic activity. To sustain their anti-tumor functions, CD8+ TILs undergo metabolic adaptation and promote FAO as an alternative energy source [159, 160]. However, excessively elevated lipid metabolism can lead to lipid peroxidation and ROS accumulation within the cell, further impairing their anti-tumor effects [161] (Fig. 4a).

Mechanism of lipid metabolic reprogramming in immune cells. A hypoxic, glucose-deficient, lipid-rich TME often educates immune cells into an immunosuppressive and pro-tumor phenotype by reprogramming their lipid metabolism. Immune cells in the TME undergo lipid metabolism reprogramming by directly absorbing excess lipids in the TME or enhancing their own lipid uptake, synthesis, and oxidation. Overexpression of PD-1 and CD36 on CD8+ TILs promotes metabolic transition by activating STAT3 or PPARs, which results in enhanced FAO (a). In Tregs cells, lipid metabolism is enhanced by CD36-PPARβ signaling, AKT-mTORC1 signaling, and SREBPs-mediated overexpression of FASN (b). TAMs can use lipids directly in TME or uptake exosomes containing LCFAs. FAO in TAMs is also enhanced by CD36-PPARs signaling (c). In DCs, NK cells, and MDSCs, CD36-PPARs signaling also plays a critical role in modulating their lipid metabolism (d–f)
CD8+ TILs often undergo metabolic transitions to adapt to the hypoxia, glucose deficiency and lipid accumulation in the TME. This transition involves converting glycolysis to FAO, maximizing their activity to maintain their anti-tumor function. For instance, in a mouse model of obesity-associated breast cancer, CD8+ TILs downregulate glycolytic activity and enhance FAO [162]. Additionally, CD8+ T cells in MC38 colorectal cancer cell line and B16 melanoma cell line-bearing mice models show an FAO increase with the upregulation of CPT1 [163,164,211]. It has been well demonstrated that increased lipid uptake and FAO in MDSCs can promote its production of immune-suppressive cytokines that inhibit cytotoxic effects of CD8+ T cells [212] (Fig. 4f).
Bioactive lipid signaling factors in TME promotes the realization of immune cells function. Specifically, in breast and colon cancer, studies have identified PGE2 as a crucial factor in activating MDSCs in TME [Full size image
Advances in targeting lipid metabolism combined with chemotherapy and targeted therapy
As described above, the dysregulated lipid metabolism in tumor cells provides energy support for tumor progression, playing an essential role in membrane synthesis and signal transduction. This shift in lipid metabolism, including the lipid uptake from the extracellular microenvironment, increased lipogenesis, and the increase in intracellular LD storage, correlates with the metastatic potential of tumor cells [224], the acquisition of stem cell-like properties [225], and the development of resistance to cancer chemotherapy [226, 227]. This may interfere with chemotherapy and targeted therapy in tumors. Therefore, aberrant lipid metabolism has become a potential target for treating drug-resistant cancer. Combination therapy can sensitize tumor cells to drugs, demonstrating a therapeutic effect superior to monotherapy [227]. Combination therapy also compensates for the insufficient efficacy of single-target lipid metabolism inhibitors. A considerable amount of preclinical research and clinical trials are currently focusing on novel strategies to enhance therapeutic effects by combining targeted lipid metabolism therapy with conventional chemotherapy and targeted therapy, even radiotherapy (Fig. 5a; Table 2).
Targeting lipid uptake
Tumor cells exhibit a propensity for lipid uptake to support biosynthesis, energy production, and lipid storage in LDs. Consequently, inhibiting lipid uptake has emerged as a promising therapeutic strategy in oncology. Considerable preclinical evidence suggests that targeting the fatty acid receptor CD36 can be effective against various types of cancer [4, 15, 228, 229]. Particularly, CD36-mediated metabolic reprogramming in breast cancer, has been linked to resistance to HER2-targeted therapies. Recent clinical studies have confirmed that higher CD36 expression correlates with poorer outcomes in early-stage HER2+ breast cancer patients undergoing trastuzumab-lapatinib therapy [230]. Furthermore, JC63.1C, an anti-CD36 monoclonal antibody, was found to resensitize lapatinib-resistant xenograft tumors to HER2-targeted therapy [231], providing a novel direction for combinatorial treatment strategies in breast cancer. In addition, studies in leukemia models showed that the FA6.152 monoclonal anti-CD36 or the CD36 inhibitor SSO, when used in conjunction with chemotherapy drugs such as Ara-C or doxorubicin, led to a significant extension in survival [232, 233]. In PDAC, CD36 siRNA notably increased the efficacy of gemcitabine treatment [234].
Inhibiting FABPs has also demonstrated promising anti-tumor effects. FABP5 inhibitors, namely SBFI-102/103, were identified as novel targets in prostate cancer and were shown to enhance the tumor-suppressing effects of paclitaxel [235, 236]. FABP4 inhibitor BMS309403, was also found to boost the efficacy of carboplatin by inhibiting tumor metastasis in in vivo ovarian cancer models [237]. In another study, a mouse model of melanoma showed that inhibiting FATP2, another critical lipid metabolism transporter, in PMN-MDSCs using lipofermata, led to enhanced tumor regression when combined with BRAF/MEK inhibitor [238]. Taken together, these findings suggest that targeting lipid uptake by inhibiting fatty acid transporters is a promising new strategy to overcome therapy resistance in cancer treatment.
Targeting lipid synthesis
Increased lipid synthesis, inclusive of fatty acids and cholesterol, is a characteristic feature of tumors. Consequently, targeting key enzymes in these pathways is emerging as a potential therapeutic strategy for cancer, particularly in conjunction with chemotherapy and targeted therapy. ACLY, a key enzyme in the production of acetyl-CoA, serving as a substrate for DNL, supports tumor growth and confers resistance to vemurafenib in melanoma treatment. In tumor-bearing mice model, although vemurafenib exerted minimal effects on tumor growth, the combination of SB-204990 and vemurafenib exhibited a significantly more suppressive effect [239]. Similarly, in vitro results suggested that combining SB-204990 could be an effective strategy for treating cisplatin-resistant ovarian cancer [240]. In addition, ACCs, which catalyze the conversion of acetyl-CoA to malonyl-CoA, the initial step in fatty acid synthesis, are upregulated in cetuximab-treated head and neck squamous cell carcinoma (HNSCC) by the continuously activated AMPK pathway, suggesting a possible mechanism refers to the cetuximab resistance. A combination of cetuximab and TOFA, an ACC inhibitor, resulted in notable growth inhibition of cetuximab-resistant HNSCC xenografts in vivo [241]. Another ACC inhibitor, ND-646, when combined with carboplatin, could also significantly inhibit tumor growth in preclinical non-small cell lung cancer (NSCLC) models [242]. Similarly, the combination of sorafenib and a liver-specific ACC inhibitor (ND-654) effectively decreased tumor proliferation in vivo [243]. However, clinical trials assessing the efficacy of targeting ACC in tumors are currently scarce. Recent clinical trials have supported the use of an ACC inhibitor (Firsocostat) for treating nonalcoholic steatohepatitis (NCT02856555, NCT03987074) [244].
Palmitic acid synthesis, another rate-limiting step in DNL, is facilitated by FASN. Deregulation in the expression and activity of FASN carries significant implications for therapeutic response. FASN inhibitors, such as the anti-obesity drug Orlistat, have shown significant anti-tumor effects in various cancers [13]. Recent studies have underscored the efficacy of combining FASN inhibitors with other treatment strategies. Preclinical evidence suggests Orlistat's ability to reverse sorafenib resistance in HCC, thus illuminating new avenues for combined HCC treatment [245, 246]. Moreover, in prostate cancer, the combination of Orlistat and radiotherapy significantly reduces the activity of the NF-κB pathway, and inhibits cancer progression [247]. Preclinical research on pancreatic cancer, NSCLC, and ovarian cancer has shown that combining Orlistat with chemotherapy and targeted treatment significantly enhances anti-tumor effects [248,249,250]. Despite Orlistat being an FDA-approved therapy, there are currently no registered clinical trials examining its role in cancer. Another FASN inhibitor, TVB-3664, exhibits synergistic therapeutic effects when combined with TKI, as shown in an HCC mouse model [251]. A separate in vivo study using a lung adenocarcinoma xenograft model demonstrated the anti-tumor effects of combining TVB-3664 with a KRAS inhibitor [252]. The PI3K pathway, frequently hyperactive in tumor, plays crucial roles in both malignant and immune cells. The co-administration of the PI3K inhibitor (CYH33) with the FASN inhibitor (C75) has been found to synergistically inhibit tumor growth while enhancing host immunity [253]. Other FASN inhibitors like TVB-3166, Fasnall, and AZ12756122 also promote enhanced anti-tumor effects in combination therapies [254, Not applicable.Availability of data and materials
Abbreviations
- FAO:
-
Fatty acid oxidation
- TME:
-
Tumor microenvironment
- CAFs:
-
Cancer-associated fibroblasts
- Tregs:
-
Regulatory T cells
- TAMs:
-
Tumor-associated macrophages
- MDSCs:
-
Myeloid-derived suppressor cells
- NK cells:
-
Natural killer cells
- TAG:
-
Triacylglycerols
- FA:
-
Fatty acid
- DNL:
-
De novo lipogenesis
- PL:
-
Phospholipid
- FATP:
-
Fatty acid transport protein
- FABP:
-
Fatty acid-binding protein
- ACLY:
-
ATP-citrate lyase
- ACSS:
-
Acetyl-CoA synthetase
- ACC:
-
Acetyl-CoA carboxylases
- FASN:
-
Fatty acid synthase
- SFA:
-
Saturated fatty acid
- MUFA:
-
Monounsaturated fatty acid
- PUFA:
-
Polyunsaturated fatty acid
- ELOVLs:
-
Elongation of very-long-chain fatty acids gene family
- SCD:
-
Stearoyl-CoA desaturase
- FADS:
-
Fatty acid desaturase
- OA:
-
Oleic acid
- LA:
-
Linoleic acid
- ALA:
-
Alpha-linolenic acid
- AA:
-
Arachidonic acid
- AdA:
-
Adrenic acid
- GLS:
-
Glutaminase
- GLUD:
-
Glutamate dehydrogenase
- IDH:
-
Isocitrate dehydrogenase
- LCFA:
-
Long-chain fatty acid
- GPAT:
-
Glycerol-3-phosphate acyltransferase
- LPA:
-
Lysophosphatidic acid
- DAG:
-
Diacylglycerol
- DGAT:
-
Diacylglycerol acyltransferase
- MVA:
-
Mevalonate
- HMGCR:
-
HMG-CoA reductase
- ABCA1:
-
ATP-binding cassette transporter A1
- LDLR:
-
Low-density lipoprotein receptor
- PC:
-
Phosphatidylcholine
- PCho:
-
Phosphocholine
- GPC:
-
Glycerophosphocholine
- ChoK:
-
Choline kinase
- PA:
-
Phosphatidic acid
- LPC:
-
Lysophosphatidylcholine
- LPCATs:
-
Lysophosphatidylcholine acyltransferase
- ATX:
-
Autotaxin
- PG:
-
Prostaglandin
- COX:
-
Cyclooxygenase
- LT:
-
Leukotriene
- LOX:
-
Lipoxygenase
- S1P:
-
Sphingosine-1-phosphate
- ACAT:
-
Acyl-CoA cholesterol acyltransferase
- ER:
-
Endoplasmic reticulum
- LD:
-
Lipid droplet
- ATGL:
-
Adipose triglyceride lipase
- HSL:
-
Hormone-sensitive lipase
- MAL:
-
Monoacylglycerol lipase
- HIG2:
-
Hypoxia-inducible gene 2
- CPT:
-
Carnitine palmitoyl transferase
- ACSL:
-
The long-chain acyl-CoA synthase enzyme family
- LPO:
-
Lipid peroxidation
- PUFA-PL:
-
PUFA-phospholipid
- MUFA-PL:
-
MUFA-phospholipid
- SREBP:
-
Sterol regulatory element-binding protein
- RTK:
-
Receptor tyrosine kinase
- PPP:
-
Pentose phosphate pathway
- HCC:
-
Hepatocellular carcinoma
- HIF:
-
Hypoxia-inducible factor
- ccRCC:
-
Clear cell renal cell carcinoma
- LRP1:
-
Low-density lipoprotein receptor-related protein 1
- FAS:
-
Fatty acid synthesis
- PPAR:
-
Peroxisome proliferator activated receptor
- HFD:
-
High-fat diet
- EVs:
-
Extracellular vesicles
- ALOX15:
-
Arachidonate lipoxygenase 15
- CD8+ TILs:
-
CD8+ tumor-infiltrating lymphocytes
- ox-LDL:
-
Oxidized low-density lipoprotein
- PDAC:
-
Pancreatic ductal adenocarcinoma
- GPX4:
-
Glutathione peroxidase 4
- MARCO:
-
Macrophage receptor for collagenous structures
- ICI:
-
Immune checkpoint inhibitor
- M-MDSC:
-
Mononuclear MDSC
- PMN-MDSC:
-
Polymorphonuclear MDSC
- NSCLC:
-
Non-small cell lung cancer
- HNSCC:
-
Neck squamous cell carcinoma
- ADT:
-
Androgen deprivation therapy
- AAD:
-
Antiangiogenic drug
- ACT:
-
Adoptive cell transfer
- PD-1:
-
Programmed cell death protein 1
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- PD-L1:
-
Programmed death-ligand 1
- PGC-1α:
-
PPARγ coactivator 1-α
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16(11):732–49.
Shang S, Liu J, Hua F. Protein acylation: mechanisms, biological functions and therapeutic targets. Signal Transduct Target Ther. 2022;7(1):396.
Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122(1):4–22.
Strickaert A, Saiselet M, Dom G, De Deken X, Dumont JE, Feron O, Sonveaux P, Maenhaut C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene. 2017;36(19):2637–42.
Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. 2016;30(5):668–81.
Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. 2020;78(6):1019–33.
Wang H, Franco F, Tsui YC, **e X, Trefny MP, Zappasodi R, Mohmood SR, Fernandez-Garcia J, Tsai CH, Schulze I, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21(3):298–308.
Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, Zhou Y, Lei J, Zhang J, Wang J, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8(5):710–21.
Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–8.
Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54(7):1561-1577 e1567.
Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, Tai LH. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer. 2019;19(1):823.
Schcolnik-Cabrera A, Chavez-Blanco A, Dominguez-Gomez G, Taja-Chayeb L, Morales-Barcenas R, Trejo-Becerril C, Perez-Cardenas E, Gonzalez-Fierro A, Duenas-Gonzalez A. Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin Investig Drugs. 2018;27(5):475–89.
Wang Y, Lu JH, Wang F, Wang YN, He MM, Wu QN, Lu YX, Yu HE, Chen ZH, Zhao Q, et al. Inhibition of fatty acid catabolism augments the efficacy of oxaliplatin-based chemotherapy in gastrointestinal cancers. Cancer Lett. 2020;473:74–89.
Ruan C, Meng Y, Song H. CD36: an emerging therapeutic target for cancer and its molecular mechanisms. J Cancer Res Clin Oncol. 2022;148(7):1551–8.
Zhang C, Liao Y, Liu P, Du Q, Liang Y, Ooi S, Qin S, He S, Yao S, Wang W. FABP5 promotes lymph node metastasis in cervical cancer by reprogramming fatty acid metabolism. Theranostics. 2020;10(15):6561–80.
Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9(1):349–65.
Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal C, Yong-Gonzales V, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 2018;8(8):1006–25.
Mendes C, Lopes-Coelho F, Ramos C, Martins F, Santos I, Rodrigues A, Silva F, Andre S, Serpa J. Unraveling FATP1, regulated by ER-beta, as a targeted breast cancer innovative therapy. Sci Rep. 2019;9(1):14107.
Wang D, Ye Q, Gu H, Chen Z. The role of lipid metabolism in tumor immune microenvironment and potential therapeutic strategies. Front Oncol. 2022;12: 984560.
Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31(1):62–76.
Khwairakpam AD, Banik K, Girisa S, Shabnam B, Shakibaei M, Fan L, Arfuso F, Monisha J, Wang H, Mao X, et al. The vital role of ATP citrate lyase in chronic diseases. J Mol Med (Berl). 2020;98(1):71–95.
Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. Acetate dependence of tumors. Cell. 2014;159(7):1591–602.
Jones SF, Infante JR. Molecular pathways: fatty acid synthase. Clin Cancer Res. 2015;21(24):5434–8.
Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, Bezwada D, Blanc L, Prideaux B, ** X, et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat Cancer. 2021;2(4):414–28.
Zhao J, Zhi Z, Wang C, **ng H, Song G, Yu X, Zhu Y, Wang X, Zhang X, Di Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol Rep. 2017;38(4):2105–15.
Ascenzi F, De Vitis C, Maugeri-Sacca M, Napoli C, Ciliberto G, Mancini R. SCD1, autophagy and cancer: implications for therapy. J Exp Clin Cancer Res. 2021;40(1):265.
Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. 2017;3(3):169–80.
** J, Byun JK, Choi YK, Park KG. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. 2023;55(4):706–15.
Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115–8.
Ippolito L, Comito G, Parri M, Iozzo M, Duatti A, Virgilio F, Lorito N, Bacci M, Pardella E, Sandrini G, et al. Lactate rewires lipid metabolism and sustains a metabolic-epigenetic axis in prostate cancer. Cancer Res. 2022;82(7):1267–82.
Perez-Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hee VF, De Saedeleer CJ, Sboarina M, Rodriguez F, Fontenille MJ, Brisson L, et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle. 2016;15(1):72–83.
Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: new perspectives and emerging mechanisms. Dev Cell. 2021;56(10):1363–93.
Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, Loda M, Kinlaw WB, Swinnen JV. Lipids and cancer: emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv Drug Deliv Rev. 2020;159:245–93.
Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016;76(8):2063–70.
Gallagher EJ, Zelenko Z, Neel BA, Antoniou IM, Rajan L, Kase N, LeRoith D. Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene. 2017;36(46):6462–71.
Kim HY, Bae SJ, Choi JW, Han S, Bae SH, Cheong JH, Jang H. Cholesterol synthesis is important for breast cancer cell tumor sphere formation and invasion. Biomedicines. 2022;10(8):1908.
Halimi H, Farjadian S. Cholesterol: an important actor on the cancer immune scene. Front Immunol. 2022;13:1057546.
Kopecka J, Trouillas P, Gasparovic AC, Gazzano E, Assaraf YG, Riganti C. Phospholipids and cholesterol: inducers of cancer multidrug resistance and therapeutic targets. Drug Resist Updat. 2020;49: 100670.
Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer. 2011;11(12):835–48.
Saito RF, Andrade LNS, Bustos SO, Chammas R. Phosphatidylcholine-derived lipid mediators: the crosstalk between cancer cells and immune cells. Front Immunol. 2022;13: 768606.
Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di Vito M, Venturini E, Glunde K, Bhujwalla ZM, Mezzanzanica D, et al. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010;70(5):2126–35.
Hernando E, Sarmentero-Estrada J, Koppie T, Belda-Iniesta C, Ramirez de Molina V, Cejas P, Ozu C, Le C, Sanchez JJ, Gonzalez-Baron M, et al. A critical role for choline kinase-alpha in the aggressiveness of bladder carcinomas. Oncogene. 2009;28(26):2425–35.
Sulciner ML, Gartung A, Gilligan MM, Serhan CN, Panigrahy D. Targeting lipid mediators in cancer biology. Cancer Metastasis Rev. 2018;37(2–3):557–72.
Shindou H, Hishikawa D, Harayama T, Yuki K, Shimizu T. Recent progress on acyl CoA: lysophospholipid acyltransferase research. J Lipid Res. 2009;50(Suppl):S46-51.
Hisano Y, Hla T. Bioactive lysolipids in cancer and angiogenesis. Pharmacol Ther. 2019;193:91–8.
Li Z, Liu H, Luo X. Lipid droplet and its implication in cancer progression. Am J Cancer Res. 2020;10(12):4112–22.
Petan T. Lipid droplets in cancer. Rev Physiol Biochem Pharmacol. 2023;185:53–86.
Moldavski O, Amen T, Levin-Zaidman S, Eisenstein M, Rogachev I, Brandis A, Kaganovich D, Schuldiner M. Lipid droplets are essential for efficient clearance of cytosolic inclusion bodies. Dev Cell. 2015;33(5):603–10.
Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, Tian Z, Cheng C, Zhang R, Puduvalli V, et al. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 2020;32(2):229-242 e228.
Wilcock DJ, Badrock AP, Wong CW, Owen R, Guerin M, Southam AD, Johnston H, Telfer BA, Fullwood P, Watson J, et al. Oxidative stress from DGAT1 oncoprotein inhibition in melanoma suppresses tumor growth when ROS defenses are also breached. Cell Rep. 2022;39(12): 110995.
Zhang R, Meng J, Yang S, Liu W, Shi L, Zeng J, Chang J, Liang B, Liu N, **ng D. Recent advances on the role of ATGL in cancer. Front Oncol. 2022;12: 944025.
Rossi T, Zamponi R, Chirico M, Pisanu ME, Iorio E, Torricelli F, Gugnoni M, Ciarrocchi A, Pistoni M. BETi enhance ATGL expression and its lipase activity to exert their antitumoral effects in triple-negative breast cancer (TNBC) cells. J Exp Clin Cancer Res. 2023;42(1):7.
Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, Liu J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. Elife. 2017. https://doi.org/10.7554/eLife.31132.
Povero D, Johnson SM, Liu J. Hypoxia, hypoxia-inducible gene 2 (HIG2)/HILPDA, and intracellular lipolysis in cancer. Cancer Lett. 2020;493:71–9.
Yin H, Li W, Mo L, Deng S, Lin W, Ma C, Luo Z, Luo C, Hong H. Adipose triglyceride lipase promotes the proliferation of colorectal cancer cells via enhancing the lipolytic pathway. J Cell Mol Med. 2021;25(8):3963–75.
Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017;2(4): e87489.
Iftikhar R, Penrose HM, King AN, Samudre JS, Collins ME, Hartono AB, Lee SB, Lau F, Baddoo M, Flemington EF, et al. Elevated ATGL in colon cancer cells and cancer stem cells promotes metabolic and tumorigenic reprogramming reinforced by obesity. Oncogenesis. 2021;10(11):82.
Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, Yang C, Batten K, Huffman KE, Liu J, et al. Fatty acid oxidation mediated by Acyl-CoA synthetase long chain 3 is required for mutant kras lung tumorigenesis. Cell Rep. 2016;16(6):1614–28.
Wang L, Li C, Song Y, Yan Z. Inhibition of carnitine palmitoyl transferase 1A-induced fatty acid oxidation suppresses cell progression in gastric cancer. Arch Biochem Biophys. 2020;696: 108664.
Nimmakayala RK, Leon F, Rachagani S, Rauth S, Nallasamy P, Marimuthu S, Shailendra GK, Chhonker YS, Chugh S, Chirravuri R, et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene. 2021;40(1):215–31.
Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30(6):478–90.
Dierge E, Debock E, Guilbaud C, Corbet C, Mignolet E, Mignard L, Bastien E, Dessy C, Larondelle Y, Feron O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021;33(8):1701-1715 e1705.
Danielli M, Perne L, Jarc Jovicic E, Petan T. Lipid droplets and polyunsaturated fatty acid trafficking: balancing life and death. Front Cell Dev Biol. 2023;11:1104725.
Martin-Perez M, Urdiroz-Urricelqui U, Bigas C, Benitah SA. The role of lipids in cancer progression and metastasis. Cell Metab. 2022;34(11):1675–99.
Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, Sell A, Wei S, Grove S, Johnson JK, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365-378 e366.
Wang C, Shi M, Ji J, Cai Q, Zhao Q, Jiang J, Liu J, Zhang H, Zhu Z, Zhang J. Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging (Albany NY). 2020;12(15):15374–91.
Luo H, Wang X, Song S, Wang Y, Dan Q, Ge H. Targeting stearoyl-coa desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology. 2022;11(1):2101769.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci. 2020;117(49):31189–97.
Dong F, Mo Z, Eid W, Courtney KC, Zha X. Akt inhibition promotes ABCA1-mediated cholesterol efflux to ApoA-I through suppressing mTORC1. PLoS ONE. 2014;9(11): e113789.
Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–35.
Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140(3):1071–83.
Li L, Pilo GM, Li X, Cigliano A, Latte G, Che L, Joseph C, Mela M, Wang C, Jiang L, et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J Hepatol. 2016;64(2):333–41.
Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15(5):273–91.
Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19(3):393–406.
Chen H, Qi Q, Wu N, Wang Y, Feng Q, ** R, Jiang L. Aspirin promotes RSL3-induced ferroptosis by suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutatnt colorectal cancer. Redox Biol. 2022;55: 102426.
Chen LL, Wang WJ. p53 regulates lipid metabolism in cancer. Int J Biol Macromol. 2021;192:45–54.
Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and metabolism. J Mol Cell Biol. 2019;11(4):284–92.
Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13(3):310–6.
Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JPT, Tschaharganeh DF, Kastenhuber ER, Barsotti AM, Culp-Hill R, et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell. 2019;176(3):564-580 e519.
Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, Tsuchihara K, Bungard D, Berger SL, Jones RG, et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013;20(4):659–68.
Wahlstrom T, Henriksson MA. Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta. 2015;1849(5):563–9.
Gouw AM, Margulis K, Liu NS, Raman SJ, Mancuso A, Toal GG, Tong L, Mosley A, Hsieh AL, Sullivan DK, et al. The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 2019;30(3):556-572 e555.
Chen J, Ding C, Chen Y, Hu W, Yu C, Peng C, Feng X, Cheng Q, Wu W, Lu Y, et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021;502:154–65.
Jia J, Che L, Cigliano A, Wang X, Peitta G, Tao J, Zhong S, Ribback S, Evert M, Chen X, et al. Pivotal role of fatty acid synthase in c-MYC Driven Hepatocarcinogenesis. Int J Mol Sci. 2020;21(22):8467.
Zhong C, Fan L, Yao F, Shi J, Fang W, Zhao H. HMGCR is necessary for the tumorigenecity of esophageal squamous cell carcinoma and is regulated by Myc. Tumour Biol. 2014;35(5):4123–9.
Zhong C, Fan L, Li Z, Yao F, Zhao H. SREBP2 is upregulated in esophageal squamous cell carcinoma and co-operates with c-Myc to regulate HMGCR expression. Mol Med Rep. 2019;20(4):3003–10.
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci. 2008;105(48):18782–7.
Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020;52(9):1496–516.
Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427–32.
Casciano JC, Perry C, Cohen-Nowak AJ, Miller KD, Vande Voorde J, Zhang Q, Chalmers S, Sandison ME, Liu Q, Hedley A, et al. MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br J Cancer. 2020;122(6):868–84.
Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–66.
Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, Shen H. Role of hypoxia-induced exosomes in tumor biology. Mol Cancer. 2018;17(1):120.
Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. 2019;120(12):1090–8.
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci. 2011;108(49):19611–6.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–4.
Liu Y, Ma Z, Zhao C, Wang Y, Wu G, **ao J, McClain CJ, Li X, Feng W. HIF-1alpha and HIF-2alpha are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1alpha-mediated fatty acid beta-oxidation. Toxicol Lett. 2014;226(2):117–23.
Ezzeddini R, Taghikhani M, Salek Farrokhi A, Somi MH, Samadi N, Esfahani A, Rasaee MJ. Downregulation of fatty acid oxidation by involvement of HIF-1alpha and PPARgamma in human gastric adenocarcinoma and related clinical significance. J Physiol Biochem. 2021;77(2):249–60.
Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun. 2017;8(1):1769.
Seo J, Jeong DW, Park JW, Lee KW, Fukuda J, Chun YS. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun Biol. 2020;3(1):638.
Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl JA, Keith B, Simon MC. HIF2alpha-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015;5(6):652–67.
Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci. 2013;110(22):8882–7.
Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, **e H, Simon MC, Kamphorst JJ. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018;24(10):2596-2605 e2595.
Mylonis I, Simos G, Paraskeva E. Hypoxia-inducible factors and the regulation of lipid metabolism. Cells. 2019;8(3):214.
Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol. 2020;82:103–26.
Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun. 2018;504(3):582–9.
Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C, Doix B, Degavre C, Guilbaud C, Petit L, et al. TGFbeta2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun. 2020;11(1):454.
Ding M, Zhang S, Guo Y, Yao J, Shen Q, Huang M, Chen W, Yu S, Zheng Y, Lin Y, et al. Tumor microenvironment acidity triggers lipid accumulation in liver cancer via scd1 activation. Mol Cancer Res. 2022;20(5):810–22.
Kondo A, Yamamoto S, Nakaki R, Shimamura T, Hamakubo T, Sakai J, Kodama T, Yoshida T, Aburatani H, Osawa T. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 2017;18(9):2228–42.
Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49(23):6449–65.
Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. 2019;15(3):139–54.
Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-associated adipocytes: key players in breast cancer progression. J Hematol Oncol. 2019;12(1):95.
Yang D, Li Y, **ng L, Tan Y, Sun J, Zeng B, **ang T, Tan J, Ren G, Wang Y. Utilization of adipocyte-derived lipids and enhanced intracellular trafficking of fatty acids contribute to breast cancer progression. Cell Commun Signal. 2018;16(1):32.
Attane C, Muller C. Drilling for oil: tumor-surrounding adipocytes fueling cancer. Trends Cancer. 2020;6(7):593–604.
Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res. 2020;80: 101055.
Tabuso M, Homer-Vanniasinkam S, Adya R, Arasaradnam RP. Role of tissue microenvironment resident adipocytes in colon cancer. World J Gastroenterol. 2017;23(32):5829–35.
Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1.
Lien EC, Vander Heiden MG. A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer. 2019;19(11):651–61.
Petrelli F, Cortellini A, Indini A, Tomasello G, Ghidini M, Nigro O, Salati M, Dottorini L, Iaculli A, Varricchio A, et al. Association of obesity with survival outcomes in patients with cancer: a systematic review and meta-analysis. JAMA Netw Open. 2021;4(3): e213520.
Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, inflammation, and cancer. Annu Rev Pathol. 2016;11:421–49.
Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, **faras ME, Akkad A, Arias E, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature. 2016;531(7592):53–8.
Kulkarni A, Bowers LW. The role of immune dysfunction in obesity-associated cancer risk, progression, and metastasis. Cell Mol Life Sci. 2021;78(7):3423–42.
Kado T, Nawaz A, Takikawa A, Usui I, Tobe K. Linkage of CD8(+) T cell exhaustion with high-fat diet-induced tumourigenesis. Sci Rep. 2019;9(1):12284.
Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, Mirsoian A, Minnar CM, Stoffel KM, Sturgill IR, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med. 2019;25(1):141–51.
Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, Liu XS, Lee YR, Fung J, Katon JM, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018;50(2):206–18.
Bousquenaud M, Fico F, Solinas G, Ruegg C, Santamaria-Martinez A. Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Res. 2018;20(1):104.
Pascual G, Dominguez D, Elosua-Bayes M, Beckedorff F, Laudanna C, Bigas C, Douillet D, Greco C, Symeonidi A, Hernandez I, et al. Dietary palmitic acid promotes a prometastatic memory via Schwann cells. Nature. 2021;599(7885):485–90.
Labbe DP, Zadra G, Yang M, Reyes JM, Lin CY, Cacciatore S, Ebot EM, Creech AL, Giunchi F, Fiorentino M, et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat Commun. 2019;10(1):4358.
Li S, Wu T, Lu YX, Wang JX, Yu FH, Yang MZ, Huang YJ, Li ZJ, Wang SL, Huang L, et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020;36: 101596.
Tutino V, De Nunzio V, Caruso MG, Veronese N, Lorusso D, Di Masi M, Benedetto ML, Notarnicola M. Elevated AA/EPA ratio represents an inflammatory biomarker in tumor tissue of metastatic colorectal cancer patients. Int J Mol Sci. 2019;20(8):2050.
Matsuoka T, Adair JE, Lih FB, Hsi LC, Rubino M, Eling TE, Tomer KB, Yashiro M, Hirakawa K, Olden K, et al. Elevated dietary linoleic acid increases gastric carcinoma cell invasion and metastasis in mice. Br J Cancer. 2010;103(8):1182–91.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99–115.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86.
Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, Zhang J, **ao Y, Sang N, Qian X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020;11(4):267.
Zhang Y, Gu Z, Wan J, Lou X, Liu S, Wang Y, Bian Y, Wang F, Li Z, Qin Z. Stearoyl-CoA Desaturase-1 dependent lipid droplets accumulation in cancer-associated fibroblasts facilitates the progression of lung cancer. Int J Biol Sci. 2022;18(16):6114–28.
Nardi F, Fitchev P, Franco OE, Ivanisevic J, Scheibler A, Hayward SW, Brendler CB, Welte MA, Crawford SE. PEDF regulates plasticity of a novel lipid-MTOC axis in prostate cancer-associated fibroblasts. J Cell Sci. 2018;131(13):jcs213579.
Hwang SH, Yang Y, Jung JH, Kim Y. Oleic acid from cancer-associated fibroblast promotes cancer cell stemness by stearoyl-CoA desaturase under glucose-deficient condition. Cancer Cell Int. 2022;22(1):404.
Peng S, Li Y, Huang M, Tang G, **e Y, Chen D, Hu Y, Yu T, Cai J, Yuan Z, et al. Metabolomics reveals that CAF-derived lipids promote colorectal cancer peritoneal metastasis by enhancing membrane fluidity. Int J Biol Sci. 2022;18(5):1912–32.
Peng S, Chen D, Cai J, Yuan Z, Huang B, Li Y, Wang H, Luo Q, Kuang Y, Liang W, et al. Enhancing cancer-associated fibroblast fatty acid catabolism within a metabolically challenging tumor microenvironment drives colon cancer peritoneal metastasis. Mol Oncol. 2021;15(5):1391–411.
Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol Cancer. 2021;20(1):154.
Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics. 2021;11(17):8322–36.
Radhakrishnan R, Ha JH, Jayaraman M, Liu J, Moxley KM, Isidoro C, Sood AK, Song YS, Dhanasekaran DN. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019;442:464–74.
Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019;9(5):617–27.
Kock A, Larsson K, Bergqvist F, Eissler N, Elfman LHM, Raouf J, Korotkova M, Johnsen JI, Jakobsson PJ, Kogner P. Inhibition of microsomal prostaglandin E synthase-1 in cancer-associated fibroblasts suppresses neuroblastoma tumor growth. EBioMedicine. 2018;32:84–92.
Elwakeel E, Bruggemann M, Wagih J, Lityagina O, Elewa MAF, Han Y, Fromel T, Popp R, Nicolas AM, Schreiber Y, et al. Disruption of prostaglandin E2 signaling in cancer-associated fibroblasts limits mammary carcinoma growth but promotes metastasis. Cancer Res. 2022;82(7):1380–95.
Weigel C, Maczis MA, Palladino END, Green CD, Maceyka M, Guo C, Wang XY, Dozmorov MG, Milstien S, Spiegel S. Sphingosine kinase 2 in stromal fibroblasts creates a hospitable tumor microenvironment in breast cancer. Cancer Res. 2023;83(4):553–67.
Neuwirt H, Bouchal J, Kharaishvili G, Ploner C, Johrer K, Pitterl F, Weber A, Klocker H, Eder IE. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun Signal. 2020;18(1):11.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, Zhang Q, Lin D, Ge S, Bai M, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19(1):43.
Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, Cirri P. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Biophys Acta. 2015;1853(12):3211–23.
Lopes-Coelho F, Andre S, Felix A, Serpa J. Breast cancer metabolic cross-talk: fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol Cell Endocrinol. 2018;462(Pt B):93–106.
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5: e10250.
Zhu GQ, Tang Z, Huang R, Qu WF, Fang Y, Yang R, Tao CY, Gao J, Wu XL, Sun HX, et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023;9(1):25.
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–41.
Luo Q, Zheng N, Jiang L, Wang T, Zhang P, Liu Y, Zheng P, Wang W, **e G, Chen L, et al. Lipid accumulation in macrophages confers protumorigenic polarization and immunity in gastric cancer. Cancer Sci. 2020;111(11):4000–11.
Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47.
DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5): e1600200.
Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, Wang Q, Yang M, Kalady MF, Qian J, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143-156 e145.
Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, Zhang Z, Li W, Lee H, Aftabizadeh M, et al. STAT3 activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. 2020;31(1):148-161 e145.
Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377-391 e379.
Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in t cells increases the number of tumor-reactive CD8(+) T Cells and facilitates anti-PD-1 therapy. Cancer Immunol Res. 2018;6(11):1375–87.
**ao L, Ma X, Ye L, Su P, **ong W, Bi E, Wang Q, **an M, Yang M, Qian J, et al. IL-9/STAT3/fatty acid oxidation-mediated lipid peroxidation contributes to Tc9 cell longevity and enhanced antitumor activity. J Clin Invest. 2022. https://doi.org/10.1172/JCI153247.
Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM, Miller BC, Juneja VR, Nguyen TH, Joshi S, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell. 2020;183(7):1848-1866 e1826.
Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, Sun Y, Li DY, Qiu HB, Wang W, et al. Fatty acid oxidation controls CD8(+) tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res. 2020;8(4):479–92.
Ma X, **ao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, Yi Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33(5):1001-1012 e1005.
Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS, Nava Lauson CB, Tiberti S, Raimondi A, Jones MA, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med. 2020. https://doi.org/10.1084/jem.20191920.
Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, Wang A, Yang M, Qian J, Dong C, et al. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J Exp Med. 2018;215(6):1555–69.
Yang W, Bai Y, **ong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531(7596):651–5.
Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, Wu K. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol. 2022;15(1):104.
Wang H, Franco F, Ho PC. Metabolic regulation of tregs in cancer: opportunities for immunotherapy. Trends Cancer. 2017;3(8):583–92.
Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, Zhang P, Panek WK, Cordero A, Han Y, et al. HIF-1alpha is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep. 2019;27(1):226-237 e224.
Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, Dhungana Y, Chapman NM, Long L, Saravia J, et al. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature. 2021;591(7849):306–11.
Kim MJ, Kim K, Park HJ, Kim GR, Hong KH, Oh JH, Son J, Park DJ, Kim D, Choi JM, et al. Deletion of PD-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat Immunol. 2023;24(1):148–61.
Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, Severa M, Rizzo F, Coccia EM, Bonacina F, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci. 2018;115(28):E6546–55.
Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499(7459):485–90.
Zhao R, Wan Q, Wang Y, Wu Y, **ao S, Li Q, Shen X, Zhuang W, Zhou Y, **a L, et al. M1-like TAMs are required for the efficacy of PD-L1/PD-1 blockades in gastric cancer. Oncoimmunology. 2020;10(1):1862520.
Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78(19):5492–503.
Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer. 2019;5(12):822–34.
Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, van Hall T, van der Burg SH. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. 2011;187(3):1157–65.
Feng Y, **ao M, Zhang Z, Cui R, Jiang X, Wang S, Bai H, Liu C, Zhang Z. Potential interaction between lysophosphatidic acid and tumor-associated macrophages in ovarian carcinoma. J Inflamm (Lond). 2020;17:23.
Linton SS, Abraham T, Liao J, Clawson GA, Butler PJ, Fox T, Kester M, Matters GL. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PLoS ONE. 2018;13(11): e0206759.
Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363(6430):989–93.
Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, Qian J, Yi Q. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020;80(7):1438–50.
Liu S, Zhang H, Li Y, Zhang Y, Bian Y, Zeng Y, Yao X, Wan J, Chen X, Li J, et al. S100A4 enhances protumor macrophage polarization by control of PPAR-gamma-dependent induction of fatty acid oxidation. J Immunother Cancer. 2021;9(6):e002548.
Yang P, Qin H, Li Y, **ao A, Zheng E, Zeng H, Su C, Luo X, Lu Q, Liao M, et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun. 2022;13(1):5782.
Xu ZZ, Xu S, Kuhlmann A, Kaech SM. The role of CD36 in macrophage lipid metabolism and function in tumor microenvironment. J Immunol. 2020;204(1):240.249.
Rabold K, Aschenbrenner A, Thiele C, Boahen CK, Schiltmans A, Smit JWA, Schultze JL, Netea MG, Adema GJ, Netea-Maier RT. Enhanced lipid biosynthesis in human tumor-induced macrophages contributes to their protumoral characteristics. J Immunother Cancer. 2020;8(2):e000638.
Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, Brunazzi EA, Vignali KM, Sun M, Stolz DB, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived interferon-gamma. Immunity. 2019;51(2):381-397 e386.
Bose D, Banerjee S, Chatterjee N, Das S, Saha M, Saha KD. Inhibition of TGF-beta induced lipid droplets switches M2 macrophages to M1 phenotype. Toxicol In Vitro. 2019;58:207–14.
Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, Schmidt F, Friedrich M, Keye J, Wan J, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. 2019;11(11): e10698.
Zhang Q, Wang H, Mao C, Sun M, Dominah G, Chen L, Zhuang Z. Fatty acid oxidation contributes to IL-1beta secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol Immunol. 2018;94:27–35.
Shu Y, Qin M, Song Y, Tang Q, Huang Y, Shen P, Lu Y. M2 polarization of tumor-associated macrophages is dependent on integrin beta3 via peroxisome proliferator-activated receptor-gamma up-regulation in breast cancer. Immunology. 2020;160(4):345–56.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9.
Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic cell metabolism and function in tumors. Trends Immunol. 2019;40(8):699–718.
Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–6.
O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23.
Oh DS, Lee HK. Autophagy protein ATG5 regulates CD36 expression and anti-tumor MHC class II antigen presentation in dendritic cells. Autophagy. 2019;15(12):2091–106.
Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–38.
Zhao F, **ao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, Liu J, Liu X, Boczkowski D, Nair S, et al. Paracrine Wnt5a-beta-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. 2018;48(1):147–60.
Mace EM, Dongre P, Hsu HT, Sinha P, James AM, Mann SS, Forbes LR, Watkin LB, Orange JS. Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol. 2014;92(3):245–55.
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by t and nk cells. Cell Metab. 2016;24(5):657–71.
Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, Hoti E, Lynch L, Geoghegan J, O’Farrelly C. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident nk cells in colorectal liver metastasis. Cancer Immunol Res. 2019;7(2):335–46.
Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19(12):1330–40.
Gong Z, Li Q, Shi J, Liu ET, Shultz LD, Ren G. Lipid-laden lung mesenchymal cells foster breast cancer metastasis via metabolic reprogramming of tumor cells and natural killer cells. Cell Metab. 2022;34(12):1960-1976 e1969.
Kobayashi T, Lam PY, Jiang H, Bednarska K, Gloury R, Murigneux V, Tay J, Jacquelot N, Li R, Tuong ZK, et al. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood. 2020;136(26):3004–17.
Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, Song JQ, Wang BB, Yuan B, Cui XL, et al. High serum levels of cholesterol increase antitumor functions of nature killer cells and reduce growth of liver tumors in mice. Gastroenterology. 2020;158(6):1713–27.
Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, Wan X. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol. 2019;10:1399.
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–64.
**ang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009;124(11):2621–33.
Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, Wang X, Zhang J, Zhang X, Zheng L, et al. A RIPK3-PGE(2) circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. 2018;78(19):5586–99.
Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, Larghi P, Rimoldi M, Tripodo C, Strauss L, et al. Tumor-derived prostaglandin E2 promotes p50 NF-kappaB-dependent differentiation of monocytic MDSCs. Cancer Res. 2020;80(13):2874–88.
Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. 2014;20(15):4096–106.
Wang Y, Jia A, Bi Y, Wang Y, Liu G. Metabolic regulation of myeloid-derived suppressor cell function in cancer. Cells. 2020;9(4):1011.
Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, Sanchez MD, Dean MJ, Rodriguez PC, Ochoa AC. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6(10): e1344804.
Wu H, Weidinger C, Schmidt F, Keye J, Friedrich M, Yerinde C, Willimsky G, Qin Z, Siegmund B, Glauben R. Oleate but not stearate induces the regulatory phenotype of myeloid suppressor cells. Sci Rep. 2017;7(1):7498.
Shi X, Pang S, Zhou J, Yan G, Sun J, Tan W. Feedback loop between fatty acid transport protein 2 and receptor interacting protein 3 pathways promotes polymorphonuclear neutrophil myeloid-derived suppressor cells-potentiated suppressive immunity in bladder cancer. Mol Biol Rep. 2022;49(12):11643–52.
Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, Wang L, Li Z, Lin L, Cao Q, et al. Regulation of ROS in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-PD-L1 tumor immunotherapy. Cell Immunol. 2021;362: 104286.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3(11):1236–47.
Munir R, Lisec J, Swinnen JV, Zaidi N. Too complex to fail? Targeting fatty acid metabolism for cancer therapy. Prog Lipid Res. 2022;85: 101143.
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, Cao Y. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76.
Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, Zhou Y, Zeng Z, Peng S, Li X, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res. 2018;37(1):118.
Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019;129(8):3006–17.
Goncalves AC, Richiardone E, Jorge J, Polonia B, Xavier CPR, Salaroglio IC, Riganti C, Vasconcelos MH, Corbet C, Sarmento-Ribeiro AB. Impact of cancer metabolism on therapy resistance–clinical implications. Drug Resist Updat. 2021;59: 100797.
Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41–5.
Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, Porter LH, Niranjan B, Whitby ST, Lo J, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med. 2019;11(478):eaau5758.
Ligorio F, Di Cosimo S, Verderio P, Ciniselli CM, Pizzamiglio S, Castagnoli L, Dugo M, Galbardi B, Salgado R, Loi S, et al. Predictive role of CD36 expression in HER2-positive breast cancer patients receiving neoadjuvant trastuzumab. J Natl Cancer Inst. 2022;114(12):1720–7.
Feng WW, Wilkins O, Bang S, Ung M, Li J, An J, Del Genio C, Canfield K, DiRenzo J, Wells W, et al. CD36-Mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep. 2019;29(11):3405-3420 e3405.
Farge T, Nakhle J, Lagarde D, Cognet G, Polley N, Castellano R, Nicolau ML, Bosc C, Sabatier M, Sahal A, et al. CD36 drives metastasis and relapse in acute myeloid leukemia. Cancer Res. 2023. https://doi.org/10.1158/0008-5472.CAN-22-3682.
Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19(1):23–37.
Kubo M, Gotoh K, Eguchi H, Kobayashi S, Iwagami Y, Tomimaru Y, Akita H, Asaoka T, Noda T, Takeda Y, et al. Impact of CD36 on chemoresistance in pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2020;27(2):610–9.
O’Sullivan SE, Kaczocha M. FABP5 as a novel molecular target in prostate cancer. Drug Discov Today. 2020. https://doi.org/10.1016/j.drudis.2020.09.018.
Carbonetti G, Converso C, Clement T, Wang C, Trotman LC, Ojima I, Kaczocha M. Docetaxel/cabazitaxel and fatty acid binding protein 5 inhibitors produce synergistic inhibition of prostate cancer growth. Prostate. 2020;80(1):88–98.
Mukherjee A, Chiang CY, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, Romero IL, Fiehn O, Lengyel E. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 2020;80(8):1748–61.
Alicea GM, Rebecca VW, Goldman AR, Fane ME, Douglass SM, Behera R, Webster MR, Kugel CH 3rd, Ecker BL, Caino MC, et al. Changes in aged fibroblast lipid metabolism induce age-dependent melanoma cell resistance to targeted therapy via the fatty acid transporter FATP2. Cancer Discov. 2020;10(9):1282–95.
Guo W, Ma J, Yang Y, Guo S, Zhang W, Zhao T, Yi X, Wang H, Wang S, Liu Y, et al. ATP-citrate lyase epigenetically potentiates oxidative phosphorylation to promote melanoma growth and adaptive resistance to MAPK inhibition. Clin Cancer Res. 2020;26(11):2725–39.
Wei X, Shi J, Lin Q, Ma X, Pang Y, Mao H, Li R, Lu W, Wang Y, Liu P. Targeting ACLY attenuates tumor growth and acquired cisplatin resistance in ovarian cancer by inhibiting the PI3K-AKT pathway and activating the AMPK-ROS pathway. Front Oncol. 2021;11: 642229.
Luo J, Hong Y, Lu Y, Qiu S, Chaganty BK, Zhang L, Wang X, Li Q, Fan Z. Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab. Cancer Lett. 2017;384:39–49.
Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22(10):1108–19.
Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, et al. Inhibition of Acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 2019;29(1):174-182 e175.
Alkhouri N, Lawitz E, Noureddin M, DeFronzo R, Shulman GI. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. 2020;29(2):135–41.
Li Y, Yang W, Zheng Y, Dai W, Ji J, Wu L, Cheng Z, Zhang J, Li J, Xu X, et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Cancer Res. 2023;42(1):6.
Shueng PW, Chan HW, Lin WC, Kuo DY, Chuang HY. Orlistat resensitizes sorafenib-resistance in hepatocellular carcinoma cells through modulating metabolism. Int J Mol Sci. 2022;23(12):6501.
Chuang HY, Lee YP, Lin WC, Lin YH, Hwang JJ. Fatty acid inhibition sensitizes androgen-dependent and -independent prostate cancer to radiotherapy via FASN/NF-kappaB pathway. Sci Rep. 2019;9(1):13284.
Papaevangelou E, Almeida GS, Box C, deSouza NM, Chung YL. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J Cancer. 2018;143(4):992–1002.
Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, Chaika NV, Yu F, Lazenby AJ, Berim L, et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017;77(20):5503–17.
Ali A, Levantini E, Teo JT, Goggi J, Clohessy JG, Wu CS, Chen L, Yang H, Krishnan I, Kocher O, et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol Med. 2018;10(3):e8313.
Wang H, Zhou Y, Xu H, Wang X, Zhang Y, Shang R, O’Farrell M, Roessler S, Sticht C, Stahl A, et al. Therapeutic efficacy of FASN inhibition in preclinical models of HCC. Hepatology. 2022;76(4):951–66.
Liu Y, Gao GF, Minna JD, Williams NS, Westover KD. Loss of wild type KRAS in KRAS(MUT) lung adenocarcinoma is associated with cancer mortality and confers sensitivity to FASN inhibitors. Lung Cancer. 2021;153:73–80.
Sun P, Zhang X, Wang RJ, Ma QY, Xu L, Wang Y, Liao HP, Wang HL, Hu LD, Kong X, et al. PI3Kalpha inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8(+)T cells and promoting fatty acid metabolism. J Immunother Cancer. 2021;9(8):e003093.
Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, Kemble G. FASN inhibition and taxane treatment combine to enhance anti-tumor efficacy in diverse xenograft tumor models through disruption of tubulin palmitoylation and microtubule organization and FASN inhibition-mediated effects on oncogenic signaling and gene expression. EBioMedicine. 2017;16:51–62.
Alwarawrah Y, Hughes P, Loiselle D, Carlson DA, Darr DB, Jordan JL, **ong J, Hunter LM, Dubois LG, Thompson JW, et al. Fasnall, a selective fasn inhibitor, shows potent anti-tumor activity in the MMTV-Neu model of HER2(+) breast cancer. Cell Chem Biol. 2016;23(6):678–88.
Polonio-Alcala E, Porta R, Ruiz-Martinez S, Vasquez-Dongo C, Relat J, Bosch-Barrera J, Ciurana J, Puig T. AZ12756122, a novel fatty acid synthase inhibitor, decreases resistance features in EGFR-TKI resistant EGFR-mutated NSCLC cell models. Biomed Pharmacother. 2022;156: 113942.
Falchook G, Infante J, Arkenau HT, Patel MR, Dean E, Borazanci E, Brenner A, Cook N, Lopez J, Pant S, et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine. 2021;34: 100797.
Kelly W, Diaz Duque AE, Michalek J, Konkel B, Caflisch L, Chen Y, Pathuri SC, Madhusudanannair Kunnuparampil V, Floyd J, Brenner A. Phase II investigation of TVB-2640 (denifanstat) with bevacizumab in patients with first relapse high-grade astrocytoma. Clin Cancer Res. 2023. https://doi.org/10.1158/1078-0432.CCR-22-2807.
Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, Furdui CM, Hegde P, Torti FM, Torti SV. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79(20):5355–66.
Pisanu ME, Maugeri-Sacca M, Fattore L, Bruschini S, De Vitis C, Tabbi E, Bellei B, Migliano E, Kovacs D, Camera E, et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J Exp Clin Cancer Res. 2018;37(1):318.
von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW, Copland JA. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res. 2013;19(9):2368–80.
Dai S, Yan Y, Xu Z, Zeng S, Qian L, Huo L, Li X, Sun L, Gong Z. SCD1 confers temozolomide resistance to human glioma cells via the Akt/GSK3beta/beta-catenin signaling axis. Front Pharmacol. 2017;8:960.
Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, Ma S, Lin CH, Copland JA, Ding J, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. 2017;67(5):979–90.
Huang Q, Wang Q, Li D, Wei X, Jia Y, Zhang Z, Ai B, Cao X, Guo T, Liao Y. Co-administration of 20(S)-protopanaxatriol (g-PPT) and EGFR-TKI overcomes EGFR-TKI resistance by decreasing SCD1 induced lipid accumulation in non-small cell lung cancer. J Exp Clin Cancer Res. 2019;38(1):129.
Pisanu ME, Noto A, De Vitis C, Morrone S, Scognamiglio G, Botti G, Venuta F, Diso D, Jakopin Z, Padula F, et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. 2017;406:93–104.
Talebi A, Dehairs J, Rambow F, Rogiers A, Nittner D, Derua R, Vanderhoydonc F, Duarte JAG, Bosisio F, Van den Eynde K, et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat Commun. 2018;9(1):2500.
Yin F, Feng F, Wang L, Wang X, Li Z, Cao Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019;10(9):672.
Dechaphunkul T, Arundon T, Raungkhajon P, Jiratrachu R, Geater SL, Dechaphunkul A. Benefits of immunonutrition in patients with head and neck cancer receiving chemoradiation: a phase II randomized, double-blind study. Clin Nutr. 2022;41(2):433–40.
Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16(11):718–31.
Hagiwara N, Watanabe M, Iizuka-Ohashi M, Yokota I, Toriyama S, Sukeno M, Tomosugi M, Sowa Y, Hongo F, Mikami K, et al. Mevalonate pathway blockage enhances the efficacy of mTOR inhibitors with the activation of retinoblastoma protein in renal cell carcinoma. Cancer Lett. 2018;431:182–9.
Kim JS, Turbov J, Rosales R, Thaete LG, Rodriguez GC. Combination simvastatin and metformin synergistically inhibits endometrial cancer cell growth. Gynecol Oncol. 2019;154(2):432–40.
Shojaei S, Koleini N, Samiei E, Aghaei M, Cole LK, Alizadeh J, Islam MI, Vosoughi AR, Albokashy M, Butterfield Y, et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020;287(5):1005–34.
Zhang Y, Liu Y, Duan J, Wang H, Zhang Y, Qiao K, Wang J. Cholesterol depletion sensitizes gallbladder cancer to cisplatin by impairing DNA damage response. Cell Cycle. 2019;18(23):3337–50.
Peng Y, He G, Tang D, **ong L, Wen Y, Miao X, Hong Z, Yao H, Chen C, Yan S, et al. Lovastatin inhibits cancer stem cells and sensitizes to chemo-and photodynamic therapy in nasopharyngeal carcinoma. J Cancer. 2017;8(9):1655–64.
Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, Kim HT, Lee JS. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2011;17(6):1553–60.
Yulian ED, Siregar NC, Bajuadji. Combination of simvastatin and FAC improves response to neoadjuvant chemotherapy in locally advanced breast cancer. Cancer Res Treat. 2021;53(4):1072–83.
Kim ST, Kang JH, Lee J, Park SH, Park JO, Park YS, Lim HY, Hwang IG, Lee SC, Park KW, et al. Simvastatin plus capecitabine-cisplatin versus placebo plus capecitabine-cisplatin in patients with previously untreated advanced gastric cancer: a double-blind randomised phase 3 study. Eur J Cancer. 2014;50(16):2822–30.
Siltari A, Riikonen J, Koskimaki J, Pakarainen T, Ettala O, Bostrom P, Seikkula H, Kotsar A, Tammela T, Helminen M, et al. Randomised double-blind phase 3 clinical study testing impact of atorvastatin on prostate cancer progression after initiation of androgen deprivation therapy: study protocol. BMJ Open. 2022;12(4): e050264.
de la Cueva A, Ramirez de Molina A, Alvarez-Ayerza N, Ramos MA, Cebrian A, Del Pulgar TG, Lacal JC. Combined 5-FU and ChoKalpha inhibitors as a new alternative therapy of colorectal cancer: evidence in human tumor-derived cell lines and mouse xenografts. PLoS ONE. 2013;8(6):e64961.
Mazarico JM, Sanchez-Arevalo Lobo VJ, Favicchio R, Greenhalf W, Costello E, Carrillo-de Santa Pau E, Marques M, Lacal JC, Aboagye E, Real FX. Choline kinase alpha (CHKalpha) as a therapeutic target in pancreatic ductal adenocarcinoma: expression, predictive value, and sensitivity to inhibitors. Mol Cancer Ther. 2016;15(2):323–33.
Abalsamo L, Spadaro F, Bozzuto G, Paris L, Cecchetti S, Lugini L, Iorio E, Molinari A, Ramoni C, Podo F. Inhibition of phosphatidylcholine-specific phospholipase C results in loss of mesenchymal traits in metastatic breast cancer cells. Breast Cancer Res. 2012;14(2):R50.
Chen Q, Hongu T, Sato T, Zhang Y, Ali W, Cavallo JA, van der Velden A, Tian H, Di Paolo G, Nieswandt B, et al. Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci Signal. 2012;5(249):ra79.
Wang D, Cabalag CS, Clemons NJ, DuBois RN. Cyclooxygenases and prostaglandins in tumor immunology and microenvironment of gastrointestinal cancer. Gastroenterology. 2021;161(6):1813–29.
Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120(1):142–56.
Yu S, Li Q, Yu Y, Cui Y, Li W, Liu T, Liu F. Activated HIF1alpha of tumor cells promotes chemoresistance development via recruiting GDF15-producing tumor-associated macrophages in gastric cancer. Cancer Immunol Immunother. 2020;69(10):1973–87.
Tan Z, **ao L, Tang M, Bai F, Li J, Li L, Shi F, Li N, Li Y, Du Q, et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics. 2018;8(9):2329–47.
Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, Hosaka K, Lim S, Wu J, He X, et al. Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab. 2018;28(1):104-117 e105.
Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P, Rhein S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond). 2007;113(4):205–12.
Reyes-Castellanos G, Abdel Hadi N, Gallardo-Arriaga S, Masoud R, Garcia J, Lac S, El Kaoutari A, Gicquel T, Planque M, Fendt SM, et al. Combining the antianginal drug perhexiline with chemotherapy induces complete pancreatic cancer regression in vivo. Iscience. 2023;26(6):106899.
Zhu J, Wu G, Song L, Cao L, Tan Z, Tang M, Li Z, Shi D, Zhang S, Li J. NKX2-8 deletion-induced reprogramming of fatty acid metabolism confers chemoresistance in epithelial ovarian cancer. EBioMedicine. 2019;43:238–52.
Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27(1):136-150 e135.
Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, Foa R, Nicolai R, Peluso G, Tafuri A. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126(16):1925–9.
Wu C, Dai C, Li X, Sun M, Chu H, Xuan Q, Yin Y, Fang C, Yang F, Jiang Z, et al. AKR1C3-dependent lipid droplet formation confers hepatocellular carcinoma cell adaptability to targeted therapy. Theranostics. 2022;12(18):7681–98.
Chen L, Ma WL, Cheng WC, Yang JC, Wang HC, Su YT, Ahmad A, Hung YC, Chang WC. Targeting lipid droplet lysophosphatidylcholine for cisplatin chemotherapy. J Cell Mol Med. 2020;24(13):7187–200.
Cotte AK, Aires V, Fredon M, Limagne E, Derangere V, Thibaudin M, Humblin E, Scagliarini A, de Barros JP, Hillon P, et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat Commun. 2018;9(1):322.
Tirinato L, Pagliari F, Di Franco S, Sogne E, Marafioti MG, Jansen J, Falqui A, Todaro M, Candeloro P, Liberale C, et al. ROS and Lipid Droplet accumulation induced by high glucose exposure in healthy colon and Colorectal Cancer Stem Cells. Genes Dis. 2020;7(4):620–35.
Senkal CE, Salama MF, Snider AJ, Allopenna JJ, Rana NA, Koller A, Hannun YA, Obeid LM. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 2017;25(3):686–97.
Hernandez-Corbacho MJ, Obeid LM. A novel role for DGATs in cancer. Adv Biol Regul. 2019;72:89–101.
Nistico C, Pagliari F, Chiarella E, Fernandes Guerreiro J, Marafioti MG, Aversa I, Genard G, Hanley R, Garcia-Calderon D, Bond HM, et al. Lipid droplet biosynthesis impairment through DGAT2 inhibition sensitizes MCF7 breast cancer cells to radiation. Int J Mol Sci. 2021;22(18):10102.
Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, Konieczny SF, Ratliff TL, Liu X, **e J, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016;35(50):6378–88.
Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, **ng B, Sun W, Ren L, Hu B, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567(7747):257–61.
Lee SS, Li J, Tai JN, Ratliff TL, Park K, Cheng JX. Avasimibe encapsulated in human serum albumin blocks cholesterol esterification for selective cancer treatment. ACS Nano. 2015;9(3):2420–32.
Li M, Yang Y, Wei J, Cun X, Lu Z, Qiu Y, Zhang Z, He Q. Enhanced chemo-immunotherapy against melanoma by inhibition of cholesterol esterification in CD8(+) T cells. Nanomedicine. 2018;14(8):2541–50.
Li J, Qu X, Tian J, Zhang JT, Cheng JX. Cholesterol esterification inhibition and gemcitabine synergistically suppress pancreatic ductal adenocarcinoma proliferation. PLoS ONE. 2018;13(2): e0193318.
Ueno G, Iwagami Y, Kobayashi S, Mitsufuji S, Yamada D, Tomimaru Y, Akita H, Asaoka T, Noda T, Gotoh K, et al. ACAT-1-regulated cholesteryl ester accumulation modulates gemcitabine resistance in biliary tract cancer. Ann Surg Oncol. 2022;29(5):2899–909.
June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. 2015;7(280):280–7.
Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175(2):313–26.
Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med. 2018;283(2):110–20.
Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, **a W, Hou J, Qiu Y, et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29(1):83–6.
Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, Lu H, Fang C, Zhang Y, Liang L, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3(4):306–17.
Cantini L, Pecci F, Hurkmans DP, Belderbos RA, Lanese A, Copparoni C, Aerts S, Cornelissen R, Dumoulin DW, Fiordoliva I, et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur J Cancer. 2021;144:41–8.
Jiang N, **e B, **ao W, Fan M, Xu S, Duan Y, Hamsafar Y, Evans AC, Huang J, Zhou W, et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat Commun. 2022;13(1):1511.
Wan H, Xu B, Zhu N, Ren B. PGC-1alpha activator-induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori. 2020;106(1):55–63.
Saibil SD, St Paul M, Laister RC, Garcia-Batres CR, Israni-Winger K, Elford AR, Grimshaw N, Robert-Tissot C, Roy DG, Jones RG, et al. Activation of peroxisome proliferator-activated receptors alpha and delta synergizes with inflammatory signals to enhance adoptive cell therapy. Cancer Res. 2019;79(3):445–51.
Kansal V, Burnham AJ, Kinney BLC, Saba NF, Paulos C, Lesinski GB, Buchwald ZS, Schmitt NC. Statin drugs enhance responses to immune checkpoint blockade in head and neck cancer models. J Immunother Cancer. 2023;11(1):e005940.
Takada K, Shimokawa M, Takamori S, Shimamatsu S, Hirai F, Tagawa T, Okamoto T, Hamatake M, Tsuchiya-Kawano Y, Otsubo K, et al. A propensity score-matched analysis of the impact of statin therapy on the outcomes of patients with non-small-cell lung cancer receiving anti-PD-1 monotherapy: a multicenter retrospective study. BMC Cancer. 2022;22(1):503.
Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, **e L, Huang Q, Li F, Li CY. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588(7839):693–8.
Schmidt NM, Wing PAC, Diniz MO, Pallett LJ, Swadling L, Harris JM, Burton AR, Jeffery-Smith A, Zakeri N, Amin OE, et al. Targeting human Acyl-CoA: cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat Commun. 2021;12(1):2814.
Giro-Perafita A, Palomeras S, Lum DH, Blancafort A, Vinas G, Oliveras G, Perez-Bueno F, Sarrats A, Welm AL, Puig T. Preclinical evaluation of fatty acid synthase and EGFR inhibition in triple-negative breast cancer. Clin Cancer Res. 2016;22(18):4687–97.
Li CF, Fang FM, Chen YY, Liu TT, Chan TC, Yu SC, Chen LT, Huang HY. Overexpressed fatty acid synthase in gastrointestinal stromal tumors: targeting a progression-associated metabolic driver enhances the antitumor effect of imatinib. Clin Cancer Res. 2017;23(16):4908–18.
Li X, Wu JB, Chung LW, Huang WC. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget. 2015;6(38):41018–32.
Shim JK, Choi S, Yoon SJ, Choi RJ, Park J, Lee EH, Cho HJ, Lee S, Teo WY, Moon JH, et al. Etomoxir, a carnitine palmitoyltransferase 1 inhibitor, combined with temozolomide reduces stemness and invasiveness in patient-derived glioblastoma tumorspheres. Cancer Cell Int. 2022;22(1):309.
Han S, Wei R, Zhang X, Jiang N, Fan M, Huang JH, **e B, Zhang L, Miao W, Butler AC, et al. CPT1A/2-mediated FAO enhancement-a metabolic target in radioresistant breast cancer. Front Oncol. 2019;9:1201.
Mao S, Ling Q, Pan J, Li F, Huang S, Ye W, Wei W, Lin X, Qian Y, Wang Y, et al. Inhibition of CPT1a as a prognostic marker can synergistically enhance the antileukemic activity of ABT199. J Transl Med. 2021;19(1):181.
Bandyopadhyay S, Li J, Traer E, Tyner JW, Zhou A, Oh ST, Cheng JX. Cholesterol esterification inhibition and imatinib treatment synergistically inhibit growth of BCR-ABL mutation-independent resistant chronic myelogenous leukemia. PLoS ONE. 2017;12(7): e0179558.
Lei J, Wang H, Zhu D, Wan Y, Yin L. Combined effects of avasimibe immunotherapy, doxorubicin chemotherapy, and metal-organic frameworks nanoparticles on breast cancer. J Cell Physiol. 2020;235(5):4814–23.
Acknowledgements
Not applicable.
Funding
This work is supported by grants from the National Natural Science Foundation of China (No.82173253), the Sichuan Province Science and Technology Support Program (No. 2022YFH0003 and No. 2023NSFSC1900), and the China Postdoctoral Science Foundation (No. 2022M712260). The funders had no role in the study design, data collection, analysis, decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
JLY and JW designed the outline. HRJ, JW, ZJW, and MJX participated in literature searches and drafted the manuscript. HRJ, JW, MJX, and BHX designed the figures and tables. JW, JLY, and KD supervised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There are no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
**, HR., Wang, J., Wang, ZJ. et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol 16, 103 (2023). https://doi.org/10.1186/s13045-023-01498-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01498-2