
Abstract
Background
Dexmedetomidine (DEX) is a selective agonist of α2-adrenergic receptors with anesthetic activity and neuroprotective benefits. However, its mechanism of action at the molecular level remains poorly defined. In this study, we investigated the protective effects of DEX on oxygen-glucose deprivation/ reperfusion (OGD/R)-induced neuronal apoptosis in PC12 cells, and evaluated its underlying mechanism(s) of neuroprotection and anti-inflammation.
Methods
An OGD/R model in PC12 cells was established. PC12 cells were cultured and divided into control, OGD/R, and OGD/R + DEX (1 μM, 10 μM, 50 μM) groups. Cell apoptosis was analyzed by flow cytometry and expression profiles were determined by qRT-PCR, western blot analysis, and enzyme linked immunosorbent assays (ELISA). The interaction between miRNA and its downstream targets was evaluated through luciferase reporter assays.
Results
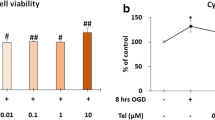
DEX significantly decreased apoptosis rates and inhibited interleukin 1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), and interleukin 6 (IL-6) release (P < 0.05). While expression of the pro-apoptotic proteins Bax and Caspase-3 was down-regulated, expression of Bcl-2 was upregulated in a dose-dependent manner (P < 0.05). Interestingly, miR-17-5p expression was down-regulated in the OGD/R group (compared to controls). Toll-like receptor 4 (TLR4), a key regulator of nuclear factor kappa-B (NF-κB) signaling, was identified as a novel target of miR-17-5p in PC12 cells. miR-17-5p expression was upregulated in the OGD/R + DEX group, suppressing TLR4 expression and reducing the secretion of proinflammatory cytokines.
Conclusion
DEX inhibits OGD/R-induced inflammation and apoptosis in PC12 cells by increasing miR-17-5p expression, downregulating TLR4, and inhibiting NF-κB signaling.
Similar content being viewed by others


Background
Stroke causes significant disabilities and cognitive impairment in afflicted individuals throughout the world [1]. The basic pathophysiology of cerebral ischemic stroke is complex, involving the interplay of autophagy, apoptosis, oxidative stress, inflammation, and energy attenuation [2, 3]. Recently, evidence has been presented highlighting the role of miRNAs in cerebral ischemia-reperfusion injury, identifying miRNAs as potential therapeutic targets [4, 5].
Dexmedetomidine (DEX) is a α2-adrenoceptor agonist that exhibits sedative, anxiolytic, and analgesic functions [6]. DEX is known to exert positive effects (in comparison with other sedatives), including mitigation of respiratory depression and hypotension, alleviation of lung and kidney damage, and decreased neuronal apoptosis [7]. DEX also has a long-term neuroprotective influence on cognitive dysfunction and brain injury [8].
The role of microRNAs (miRs), short non-coding RNA molecules, is to bind to mRNAs and inhibit the expression of target genes. The downregulation of miRs in neuronal cells is intricately linked to neurodegenerative disease [9]. Approximately 70% of all known miRs are expressed in the brain (either locally or tissue wide), and these are critical to the functionality of the nervous system [10]. Oxygen-glucose deprivation/reperfusion (OGD/R) miRs are reported to suppress apoptosis of develo** hippocampal astrocytes in rodents, thus affording protection against hepatic ischemia/ reperfusion injury [11, 12]. An interaction between miR-223-3p and TIAL1 has been demonstrated to contribute to the neuroprotective effects of DEX in hippocampal neuronal cells in vitro [13]. Hence, DEX may regulate OGD/R-induced inflammation and apoptosis through miRs. Hao et al. (2017) report that miR-17-5p is pro-apoptotic, and that miR-17-5p overexpression induces neuronal death and apoptosis [19]. DEX preconditioning is reported to protect the heart from apoptosis following ischemic/ reperfusion injury in a diabetic rat model (both in vivo and in vitro) by activating PI3K/ Akt signaling [20]. Furthermore, DEX preconditioning protects the heart from apoptosis in ischemic/ reperfusion injury in diabetic rats by activating PI3K/ Akt signaling in vivo and in vitro [21]. However, the mechanisms by which DEX regulates these effects remain to be elucidated.
miRNAs participate in a range of essential biological processes, including neuronal apoptosis during ischemic stroke and nervous system dysfunction [22]. Here, we explored the underlying biological mechanism of DEX attenuation of OGD/R-induced neurotoxicity, and we investigated the involvement of miR-17-5p and potential molecular factors. miR-17-5p is significantly upregulated in the early stage of cerebral ischemia-reperfusion injury (within 4 h). Moreover, onset of miR-17-5p upregulation is earlier than the observed changes in urea nitrogen level and neutrophil gelatinase-related lipocalin (NGAL) concentration [23]. miR-17-5p is known to be induced by p53 and to protect from renal ischemia-reperfusion injury by targeting death receptor 6 [24]. In addition, miR-17-5p is down-regulated by the Act A/ Smads signaling loop, thus enhancing the neuroprotective effect after ischemic injury [25]. Although these effects of miR-17-5p may modulate the therapeutic efficacy of DEX on OGD/R-induced neurotoxicity, no reports to this effect have been published thus far.
TLR4 belongs to the Toll-like receptor family. These innate pattern recognition receptors mediate the host response to pathogen infection [26]. TLR4 activation promotes the production of inflammatory cytokines, such as IL-1β, TNF-α, and IL-6 [27]. Aberrant IL-1β and IL-6 responses induced by TLR4 have been observed in patients. TLR4 mRNA is reported to have a binding site for miR-17-5p [28]. Thus, miR-17-5p may regulate inflammation induced by oxygen and glucose deprivation/ reperfusion via TLR4/ NF-κB [29].
In our present study, we report that miR-17-5p was downregulated in the OGD/R group, and that miR-17-5p mediated OGD/R-induced inflammation and apoptosis. DEX treatment increased miR-17-5p expression in a dose-dependent manner in PC12 cells. Moreover, miR-17-5p overexpression suppressed the inflammatory response by inhibiting NF-κB. Conversely, miR-17-5p downregulation produced the opposite phenotype. To explore the mechanisms underlying these effects, the TargetScan V7.2 database was employed to identify miR-17-5p regulated genes. miR-17-5p was predicted to bind to TLR4 mRNA. Our results reveal that miR-17-5p levels were negatively correlated with TLR4 levels, and that miR-17-5p binding to the 3′-UTR of TLR4 suppressed expression in a luciferase gene reporter assay. However, the role of miR-17-5p remained undefined.
To further investigate the role of miR-17-5p in OGD/R-induced inflammation and apoptosis in PC12 cells, miR-17-5p mimic and miR-17-5p inhibitor were separately transfected into each group. miR-17-5p mimic was observed to inhibit OGD/R-induced inflammation and apoptosis. Moreover, we identified NF-κB signaling as a potential mediator of miR-17-5p inhibition, and we subsequently demonstrated that miR-17-5p mimic could inhibit TLR4/ NF-κB signaling. For many drugs such as soy isoflavones and genistein, phosphorylation of IκBα and P65 is necessary for their neuroprotective effects [30]. From a consideration of these results, we propose that DEX upregulates miR-17-5p, and that miR-17-5p inhibits NF-κB subsequently reducing OGD/R-induced inflammation and apoptosis. In addition, we observed that inhibition of OGD/R-induced inflammation and apoptosis was suppressed following TLR4 overexpression or miR-17-5p silencing, which suggests that DEX attenuates oxygen-glucose deprivation/ reperfusion-induced inflammation and apoptosis in PC12 cells through an effect on the miR-17-5p/ TLR4/ NF-κB axis. Together, our data demonstrate the potential of DEX as a novel intervention strategy for cerebral ischemia-reperfusion injury. These findings require further verification in human stroke patients.
Our study had several limitations worth noting. First, the optimal concentration of DEX in vivo was not investigated. Therefore, DEX dosage should be further investigated in in vivo experiments. Second, DEX has only been observed thus far to up-regulate the expression of miR-17-5p in PC12 cells. Further studies are required to confirm these effects in vivo. On the side, in this study, DEX mainly exerts its protective effects through its anti-inflammatory activity, which is an ancillary activity compared to its main property (hypnotic drug). Would glucocorticoids, the reference anti-inflammatory drugs, provide the same protection while avoiding undesired central effects? Several studies in recent years have reported on this, Wang et al. [31] found knockout in studies glucocorticoid-regulated kinase 1 (SGK1) knockdown upregulated beclin-1 and LC-3 expression mediated by Intracarotid cold saline infusion (ICSI), This suggests that ICSI has a neuroprotective effect on ischemic stroke after reperfusion through up-regulation of SGK1 and inhibition of autophagy. The role of glucocorticoid signaling was also reported in the study of Perović et al. [32], They found Food restriction (FR) applied prior to TBI significantly changes p-GR levels, and it’s transcriptional activity during the recovery period after TBI. Moreover, as a pretreatment, FR modulates other protective factors in response to TBI, such as 11β-HSD1, NF-κB (p65) and HSP70 that act in parallel with GR in it’s anti-inflammatory and neuroprotective effects in the rat model of brain injury. That’s what we’re going to do next. Conclusion.
DEX inhibits OGD/R-induced inflammation and apoptosis in PC12 cells by increasing miR-17-5p expression, downregulating TLR4, and inhibiting NF-κB signaling. These results preliminarily explain the neuroprotective mechanism of DEX in ischemic stroke, and provide a direction for further searching for therapeutic targets for ischemic stroke.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
The authors declare that there was no conflict of interest in this paper.
Abbreviations
- DEX:
-
Dexmedetomidine
- ELISA:
-
Enzyme Linked Immunosorbent Assays
- miRs:
-
microRNAs
- mimic NC:
-
Mimic Negative Controls
- inhibitor NC:
-
Inhibitor Negative Controls
- IL-1β:
-
Interleukin 1 beta
- TNF-α:
-
Tumor Necrosis Factor-alpha
- IL-6:
-
Interleukin 6
- WT:
-
Wild-type
- TBST:
-
Tris Buffered Saline containing Tween 20
- SD:
-
Standard Deviation of the mean
- Bax:
-
Bcl-2 associated X protein
- Caspase-3:
-
Cysteinyl aspartate specific proteinase-3
References
Huang XP, Ding H, Yang XQ, et al. Synergism and mechanism of Astragaloside IV combined with Ginsenoside Rg1 against autophagic injury of PC12 cells induced by oxygen glucose deprivation/reoxygenation. Biomed Pharmacother. 2017;89:124–34.
Papanagiotou P, White CJ. Endovascular reperfusion strategies for acute stroke. JACC Cardiovasc Interv. 2016;9:307–17. https://doi.org/10.1016/j.jcin.2015.11.014.
Schaller B, Graf R. Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab. 2004;24:351–71.
Ogata K, Sumida K, Miyata K, Kushida M, Kuwamura M, Yamate J. Circulating miR-9* and miR-384-5p as potential indicators for trimethyltin-induced neurotoxicity. Toxicol Pathol. 2015;43:198–208.
Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7(12):911–20.
Alam A, Suen KC, Hana Z, Sanders RD, Maze M, Ma D. Neuroprotection and neurotoxicity in the develo** brain: an update on the effects of dexmedetomidine and xenon. Neurotoxicol Teratol. 2017;60:102–16.
Endesfelder S, Makki H, von Haefen C, Spies CD, Bührer C, Sifringer M. Neuroprotective effects of dexmedetomidine against hyperoxia-induced injury in the develo** rat brain. PLoS One. 2017;12:e0171498.
Degos V, Charpentier TL, Chhor V, et al. Neuroprotective effects of dexmedetomidine against glutamate agonist-induced neuronal cell death are related to increased astrocyte brain-derived neurotrophic factor expression. Anesthesiology. 2013;118:1123–32.
Ding L, Zhang H, Mi W, et al. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2015;40:129–35.
Paeschke N, von Haefen C, Endesfelder S, Sifringer M, Spies CD. Dexmedetomidine Prevents Lipopolysaccharide-Induced MicroRNA Expression in the Adult Rat Brain. Int J Mol Sci. 2017;18:1830.
Zhang L, Dong LY, Li YJ, Hong Z, Wei WS. The microRNA miR-181c controls microglia-mediated neuronal apoptosis by suppressing tumor necrosis factor. J Neuroinflammation. 2012;9:211.
Sun ZZ, Lv ZY, Tian WJ, Yang Y. MicroRNA-132 protects hippocampal neurons against oxygen-glucose deprivation-induced apoptosis. Int J Immunopathol Pharmacol. 2017;30:253–63.
Sun WC, Liang ZD, Pei L. Propofol-induced rno-miR-665 targets BCL2L1 and influences apoptosis in rodent develo** hippocampal astrocytes. Neurotoxicology. 2015;51:87–95.
Hao W, Zhao ZH, Meng QT, Tie ME, Lei SQ, **a ZY. Propofol protects against hepatic ischemia/reperfusion injury via miR-133a-5p regulating the expression of MAPK6. Cell Biol Int. 2017;41:495–504.
Wang Q, Yu H, Yu H, Ma M, Ma Y, Li R. miR-223-3p/TIAL1 interaction is involved in the mechanisms associated with the neuroprotective effects of dexmedetomidine on hippocampal neuronal cells in vitro. Mol Med Rep. 2019;19:805–12.
Wang JX, Jia XJ, Liu Y, et al. Silencing of miR-17-5p suppresses cell proliferation and promotes cell apoptosis by directly targeting PIK3R1 in laryngeal squamous cell carcinoma. Cancer Cell Int. 2020;20:14.
An JH, Chen ZY, Ma QL, Wang HJ, Zhang JQ, Shi FW. LncRNA SNHG16 promoted proliferation and inflammatory response of macrophages through miR-17-5p/NF-κB signaling pathway in patients with atherosclerosis. Eur Rev Med Pharmacol Sci. 2019;23:8665–77.
Wang M, Li YJ, Ding Y, et al. Silibinin prevents autophagic cell death upon oxidative stress in cortical neurons and cerebral ischemia-reperfusion injury. Mol Neurobiol. 2016;53:932–43.
Li C, Liu Y, Tang P, et al. Hydrogen sulfide prevents OGD/R-induced apoptosis by suppressing the phosphorylation of p38 and secretion of IL-6 in PC12 cells. Neuroreport. 2016;27:230–4.
Chang JH, ** M, Liu JT. Dexmedetomidine pretreatment protects the heart against apoptosis in ischemia/reperfusion injury in diabetic rats by activating PI3K/Akt signaling in vivo and in vitro [J]. Biomed Pharmacother. 2020;127:110188.
Sun W, Zhao J, Li C. Dexmedetomidine provides protection against hippocampal neuron apoptosis and cognitive impairment in mice with Alzheimer’s disease by mediating the miR-129/YAP1/JAG1 axis [J]. Mol Neurobiol. 2020;57(12):5044–55.
Liu R, Zhong X, Zeng J, et al. 3′-Daidzein sulfonate sodium inhibits neuronal apoptosis induced by cerebral ischemia-reperfusion. Int J Mol Med. 2017;39:1021–8.
Ma L, Wu K, Liu K, et al. Changes of miRNA-17-5p, miRNA-21 and miRNA-106a level during rat kidney ischemia-reperfusion injury [J]. Zhonghua Yi Xue Za Zhi. 2015;95(19):1488–92.
Hao J, Wei Q, Mei S, et al. Induction of microRNA-17-5p by p53 protects against renal ischemia-reperfusion injury by targeting death receptor 6[J]. Kidney Int. 2017;91(1):106–18.
Wang JQ, Dong Y, Li SJ, et al. Knockdown of microRNA-17-5p enhances the neuroprotective effect of Act A/Smads signal loop after ischemic injury [J]. Neurochem Res. 2019;44(8):1807–17.
Liu B, Li F, Shi J, Yang D, Deng Y, Gong Q. Gastrodin ameliorates subacute phase cerebral ischemia-reperfusion injury by inhibiting inflammation and apoptosis in rats. Mol Med Rep. 2016;14:4144–52.
Ma D, Hossain M, Rajakumaraswamy N, et al. Dexmedetomidine produces its neuroprotective effect via the alpha 2A-adrenoceptor subtype. Eur J Pharmacol. 2004;502:87–97.
Ji ZR, Xue WL, Zhang L. Schisandrin B attenuates inflammation in LPS-induced sepsis through miR-17-5p downregulating TLR4[J]. Inflammation. 2019;42(2):731–9.
Suo L, Wang M. Dexmedetomidine attenuates oxygen-glucose deprivation/reperfusion-induced inflammation through the miR-17-5p/TLR4/NF-κB axis [J]; 2020.
Huang R, Chen Y, Yu AC, Hertz L. Dexmedetomidine-induced stimulation of glutamine oxidation in astrocytes: a possible mechanism for its neuroprotective activity. J Cereb Blood Flow Metab. 2000;20:895–8.
Wang D, Huang Z, Li L, et al. Intracarotid cold saline infusion contributes to neuroprotection in MCAO-induced ischemic stroke in rats via serum and glucocorticoid-regulated kinase 1[J]. Mol Med Rep. 2019;20(4):3942–50.
Perović M, Jović M, Todorović S, et al. Neuroprotective effects of food restriction in a rat model of traumatic brain injury - the role of glucocorticoid signaling. Nutr Neurosci. 2022;25(3):537–49. Epub 2020.
Acknowledgements
None to declare.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
LS: Supervision; Methodology; Investigation; Data Curation; Writing – Original Draft; Writing – Review & Editing; Visualization. MW: Conceptualization; Resources; Data Curation; Writing – Review & Editing; Project Administration. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All protocols used during our study followed the requirements of the Animal Experiment Center of the Institute of Radiation Medicine of the Chinese Academy of Medical Sciences.
Consent for publication
Not applicable.
Competing interests
No potential conflicts of interest relevant to this article are reported.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Suo, L., Wang, M. Dexmedetomidine attenuates oxygen-glucose deprivation/ reperfusion-induced inflammation through the miR-17-5p/ TLR4/ NF-κB axis. BMC Anesthesiol 22, 126 (2022). https://doi.org/10.1186/s12871-022-01661-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12871-022-01661-1