
Abstract
Background
The mechanisms underlying rice root responses to drought during the early developmental stages are yet unknown.
Results
This study aimed to determine metabolic differences in IR64, a shallow-rooting, drought-susceptible genotype, and Azucena, a drought-tolerant and deep-rooting genotype under drought stress. The morphological evaluation revealed that Azucena might evade water stress by increasing the lateral root system growth, the root surface area, and length to access water. At the same time, IR64 may rely mainly on cell wall thickening to tolerate stress. Furthermore, significant differences were observed in 49 metabolites in IR64 and 80 metabolites in Azucena, for which most metabolites were implicated in secondary metabolism, amino acid metabolism, nucleotide acid metabolism and sugar and sugar alcohol metabolism. Among these metabolites, a significant positive correlation was found between allantoin, galactaric acid, gluconic acid, glucose, and drought tolerance. These metabolites may serve as markers of drought tolerance in genotype screening programs. Based on corresponding biological pathways analysis of the differentially abundant metabolites (DAMs), biosynthesis of alkaloid-derivatives of the shikimate pathway, fatty acid biosynthesis, purine metabolism, TCA cycle and amino acid biosynthesis were the most statistically enriched biological pathway in Azucena in drought response. However, in IR64, the differentially abundant metabolites of starch and sucrose metabolism were the most statistically enriched biological pathways.
Conclusion
Metabolic marker candidates for drought tolerance were identified in both genotypes. Thus, these markers that were experimentally determined in distinct metabolic pathways can be used for the development or selection of drought-tolerant rice genotypes.
Similar content being viewed by others

Introduction
Drought is a major abiotic stress that restricts crop growth, development and yield and thus has turned into a grave threat to universal food security [1]. In addition, global climate change, particularly high temperatures and erratic rainfall patterns, combined with a growing world population, is placing tremendous stress on food security and sustainability. These challenging conditions can be overcome through breeding programs to develop drought-resistant crops [2, 3]. The combined effect of drought and other abiotic stresses can reduce potential crop production by more than 50%. According to modeling simulations, drought-affected crop** regions could quadruple by the end of the twenty-first century [4]. In response to these conditions, rice employs various adaptive methods, such as building up various osmoprotectants or solutes and changes in the direction of plant growth to avoid drought [5]. Roots are the plant’s main organs that anchor it in the soil and are required for nutrition and water absorption. Favorable responses of plants to water stress is dependent on the roots’ capacity to maintain growth (i.e., modifying the root traits such as depth, density, and root angles) and maintain/increase root hydraulic conductivity [6,7,8]. The ability of roots to tolerate water deficiency depends on their ability to maintain adequate carbohydrate metabolism, cell wall protein composition, osmotic potential, and metabolites involved in the oxidative stress response [9, 10]. Most research has been focused on improving features in above-ground tissues to tolerate these pressures, but roots (the ‘hidden half’ of a plant’s architecture) remain an underutilized source of crop development [11]. Root System Architecture (RSA) is critical for improving nutrient and water uptake and maintaining crop yield under optimal and drought conditions [12]. Extensive root systems can help plants overcome drought, which is influenced by growth angle, root thickness, and length [13, 2). The score plot of the first two PCs is shown in Fig. 1A. The majority of the data fell inside the 92% confidence interval (Hotelling T2 ellipse). The PCA results for the four cluster samples showed that there was a clear distinction between the control (c) and treated samples (drought-stressed), while no clear difference was seen between the two genotypes (Fig. 1A). To confirm this trend, three of the whole competitive groups were examined by PCA, yielding similar results. The PCA models yielded two, two, and three PCs, respectively, for comparing AZs vs. AZc, IRs vs. IRc, and AZs vs. IRs samples, respectively. The R2 X and Q2 (goodness of prediction) values are shown in Table 2 and indicate that the differences between groups could be predictably explained by all the models. However, no distinct limit could be seen between the two groups’ PCA score plots (Table 2). PLS-DA is a supervised method that categorizes the observations into groups that yield the largest predicted indicator variable. The obtained data resulted in two PCs (R2 X = 0.586, R2 Y = 0.512, Q2 = 0.471) between the four cluster, and enhanced the classification between these groups in the score plot (Fig. 1B). It led to better modeling and prediction results with two PCs, when the data were examined with only the control and the treated-AZ or treated-IR64 (R2 Y > 0.9, Q2 > 0.7) (Table 2) As shown in Table 2, the treated AZ and IR samples could be separated with two PCs in spite of the slight overlap in the PLS-DA score plot (R2 Y > 0.5, Q2 > 0.08), indicating intrinsic metabolic differences between these two genotypes in the treated conditions.

Principal component analysis (PCA) and Partial least squares-discriminate analysis (PLS-DA) score plots of metabolic profiles in rice roots under drought stress. A PCA score plot for Azucena normal (red), IR64 normal (blue), Azucena-treated (yellow) and IR64-treated (green) samples, B PLS-DA score plot for Azucena normal (red), IR64 normal (blue), Azucena-treated (yellow) and IR64-treated (green) samples
Overview of Azucena and IR64 root-tips metabolome under control and drought stress conditions
The line plots of the X-loadings of the first component of the PLS-DA pairwise comparison models were used to identify the primary altered metabolites. Based on the parameter VIP > 1, p-value ≤ 0.01, log fold change (log FC > 2.0), and Kruskal–Wallis ANOVA, a total of 103 drought responsive metabolites with significant differences were identified (Table S2 and S3). The most significant metabolites had variable importance in the projection (VIP) values greater than 1, which was reported to explain the responses [30]. The VIP values for metabolites categorized by superclass are shown in Fig. 2A. Amino acid and organic acid groups were most abundant. For example, GABA and aspartic acid had a VIP value of 20.52 and 18.30, although the average value of the exclusive VIP was 5.63. Organic acid (16%), amino acid (12.2%), polyphenols (12.8%), nucleic acid derivatives (8.3%) and unclassified (others, 7.1%) were the most common differentially abundant metabolites (Fig. 2B).

Classification of the differentially abundant metabolites and their variable importance in the projection (VIP) distribution in IR64 and Azucena in root tips. A VIP distribution in each metabolite superclass as a scatter plot. The average mean of the differentially abundant metabolites is shown in the red dashed line (B) A pie chart depicting the proportion of each metabolite in the superclass
Compared to the control condition, the levels of 56 metabolites increased and 24 metabolites decreased in the Azucena root-tips in response to drought stress. In contrast, in IR64, the levels of 30 metabolites increased, and 19 decreased in response to drought stress (Fig. 3A). A cross-comparison of the differentially abundant metabolites between genotypes showed that number of accumulated metabolites were nearly two times higher in the root tips of the tolerant genotype (53 metabolites) than the sensitive one (22 metabolites), and of which 27 metabolites were commonly altered between the two genotypes in response to drought stress (Fig. 3B). The 25 most differentially abundant metabolites are shown in Fig. 4. The log2 fold change values and VIP score for these metabolites are mentioned in Table S2 & Table S3. As shown in the Fig. 4, the levels of amino acids including aspartic acid and glutamic acid, nucleotides namely thymine and guanine increased in both genotypes under drought stress. Interestingly, glycine, phenylalanine, threonine, isoleucine, and GABA were accumulated in IR64, while there was no significant difference in the levels of these metabolites in Azucena under drought stress. Likewise, there is a considerable increase in the levels of TCA cycle intermediates comprising of malic acid, isocitric acid, succinic acid and fumaric acid, and sugar and sugar alcohol such as sorbitol, mannitol, galactinol, myo-inositol, D-raffinose, sucrose, ribose and trehalose. In particular, the levels of several metabolites involved in secondary metabolism comprising of genistein, vanillin, scopoletin, conifery aldehyde, farnesyl pyrophosphate, betaine, cyanidin 3-O-rutinoside 5-O-beta-D-glucoside, and syringic acid showed the highest levels in the drought tolerant cultivar Azucena.

Overview of differentially abundant metabolites (DAMs) between the Azucena and IR64. A Up- and down-regulated metabolites of Azucena and IR64 in response to drought stress. B Venn diagram showing the overlap between DAMs responsive to drought stress

A Bar graph of 25 differentially abundant metabolites in Azucena root tips. VIP values are in a blue column and the red columns represent log2 (fold change, FC) values. B Bar graph of 25 differentially abundant metabolites in IR64 root tips. VIP values are in a blue column and the red columns represent log2 (fold change, FC) values
Identification of potential association between metabolites and observed root traits
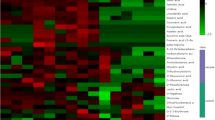
Metabolite content was determined in the root tips of control and drought-stressed plants from two different genotypes. The hierarchical clustering for both metabolites and samples (genotypes × conditions) is shown in Fig. 5. Clustering of the samples showed complete separation of the metabolite pattern between the control and drought-treated samples. Thus, drought treatment was the main source of variance in the data, indicating a complete change in metabolism under stress conditions in both genotypes. Figure 5 shows the metabolites that changed significantly under drought stress; glutamic acid, aspartic acid, proline, glucose-6-phosphate, and thymine were among the predominant metabolites that increased in response to drought stress, whereas metabolites belonging to quinic acid and ribonic acid (lowest group in Fig. 5) decreased under drought stress.

Metabolite response to drought differs between the two rice cultivars. Hierarchical clustering and heatmap of metabolite levels in root tip of IRc: IR64 genotype under control condition, IRs: IR64 genotype under drought stress conditions, AZc: Azucena genotype under control conditions, AZs: Azucena genotype under drought stress conditions
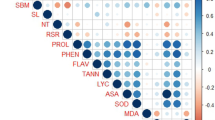
The potential correlation between the abundant metabolites and the root phenotype were tested by analyzing the correlations of expression of the metabolite levels with phenotypic traits. We rely on the prediction marker for high concentrations since high amounts of a metabolite can be detected more accurately than low concentrations or their absence. High metabolite concentrations in tolerant cultivars are indicated by significant positive correlations of metabolites with phenotypic traits, whereas high concentrations in susceptible cultivars are indicated by significant negative correlations. In a positive correlation, the metabolite would be a tolerance marker because its higher concentrations would contribute to the tolerance. Metabolites with negative correlation are sensitivity markers. Most of the significant correlations between metabolite levels and root phenotypic parameters were found to be positive under drought stress (Fig. 6). Positive correlations were observed for the concentration of the trehalose, proline, succinic acid, tryptophan, salicylic acid, sucrose, fructose lysine and mannitol. Higher concentrations of these metabolites were associated with the number of tips, root length (cm), surface area (cm2), and root diameter. In tolerant plants as opposed to sensitive plants, concentrations of these metabolites were higher during drought stress.

Correlation of root phenotypic data with metabolite levels. Correlation coefficients for selected metabolites with significant (p≤0.05) positive (blue) or negative (red) correlation between concentration metabolite levels with root phenotypic data under drought or control conditions. Data of root tip of two cultivars (Azucena and IR64) grown in two conditions. Mean values of three to five replicates per cultivar and condition were correlated. ARA (Analyzed Region Area (cm2)); ARW (Analyzed Region Width (cm)); ARH (Analyzed Region Height (cm)); RL (Root Length (cm)); SA (Surface Area (cm2)); N.Tip ( number of tips), LR ( lateral root (%)), RD (root diameter (cm)
However, for most of these metabolites, no correlations were found between concentrations under control conditions and root performance under drought. In contrast, concentrations of ribose under control conditions correlated positively with performance under drought and since there was a positive correlation between concentrations and root traits performance under control conditions also, ribose concentrations appear to be related to rate of root growth rather than drought tolerance. Gallic acid and ascorbic acid made better candidates for drought markers because their concentrations exhibited positive correlation with root performance only under drought stress conditions. Negative correlations were found for concentrations of leucine, isoleucine, pyroglutamic acid, phenylalanine, and glycerol. Higher concentrations of these metabolites were associated with decreased root length, surface area, and number of root tips. However, GABA concentration was strongly correlated with root diameter. The levels were 10 to 100 times higher in the sensitive genotype than in the tolerant genotype.
Functional annotation of DAMs
The differentially altered metabolites were functionally categorized based on the DAMs in Azucena and IR64, respectively, according to the KEGG database (www.kegg.jp/kegg/kegg1.html). When the control and treated plants were compared, it was observed that drought stress significantly altered the relative abundance of the levels of several metabolite classes. The most represented categories were organic acid compounds, amino acids, polyphenols, flavonoids, and sugars. Between the Azucena and IR64 genotypes, there was a significant variation in the proportion of organic acid compounds and biosynthesis of carbohydrates and amino acids. The metabolites in Azucena were mainly associated with amino acid biosynthesis and the TCA cycle, aminoacyl-tRNA biosynthesis, and fatty acid biosynthesis (Fig. 7) while metabolites in the IR64 genotype were mainly related to phenylalanine and galactose metabolism (p < 0.05) (Fig. 8). Increase in sucrose, glucose, tryptophan, and proline in the metabolic pathways of Azucena, may have a substantial impact on how resistant Azucena is to drought. Drought stress can have severe consequences for phenylalanine production in IR64. The high GABA expression together with its negative string correlation with root traits also indicates that IR64 restricts root elongation. Candidate DAMs demonstrating important functions or variant-specific expression profiles in Azucena and IR64 are listed in Table S2 and S3, and their relationships to major functional categories are shown in Fig. 9. A total of 103 DAMs were enriched in 30 metabolic pathways, which were most strongly represented by starch and sucrose metabolism, amino acid biosynthesis, secondary metabolite biosynthesis, purine metabolism, and fatty acid biosynthesis.

KEGG pathway classification of the differentially abundant metabolites under drought stress in Azucena (www.kegg.jp/kegg/kegg1.html)

KEGG pathway classification of the differentially abundant metabolites under drought stress in IR64 (www.kegg.jp/kegg/kegg1.html)

Primary metabolism responses in two rice genotypes (Azucena and IR64) after drought stress. Colors depict the relative accumulation levels of each metabolite: white (no significance), red (increase) and blue (decrease)
Expression of genes involved in drought stress response
Potential marker genes involved in sugar metabolism, root growth and elongation were identified in our previous transcriptome studies on two rice genotypes with different drought tolerance [31, 32]. Thirteen genes were selected as candidate markers, and their expression levels were measured by qRT-PCR in the roots of the two genotypes grown under control and drought stress conditions. It was found that the gene Ethylene response factor (ERF35) was significantly expressed in the Azucena genotype (5.63-fold), whereas it was less expressed in the IR64 genotype (3.56-fold). Gene expression of serine/threonine kinase (SnRK2), IAA19, Trehalose -phosphate phosphate1 (OsTPP1), SUCROSE TRANSPORTER 5 (SUT5), dehydration responsive element binding protein (DREB) increased (upregulated) in Azucena by 4.86-fold, 5.38-fold, 5.90-fold, 8.45-fold and 6.75-fold, respectively, while they had no detectable change in IR64 (Fig. 10). Primer sequences and more details about genes can be found in Table S6. To find the interrelation between expression levels of selected genes and metabolites, the correlation analysis was done. The connections between metabolite levels and expression levels were in the positive rather than in the negative direction indicating a co-regulation at both the transcriptional and metabolite levels. For instance, there was a significant positive correlation between the level of proline and expression of the pyrroline-5-carboxylate reductase (P5CR) which in involved in proline metabolism (Fig. 11). Further positive connections were found between PRP5, glycine- and proline-rich protein 3 (OsGPRP3), trehalose phosphate phosphate1 (TPP1), sucrose transport protein (SUT5), and auxin-responsive protein IAA19 (IAA19) with trehalose, sucrose, and tryptophan, which were present at high levels under drought condition (Fig. 11). Thus, the high expression of these genes indicates drought tolerance under drought stress conditions.

Validations of selected genes using qRT-PCR in root tip zone of both genotypes, Azucena and IR64, in response to water stress

Correlation of the expression level of selected genes with the abundance of metabolites. Correlation coefficients for selected metabolites with significant (p ≤ 0.05) positive (blue) or negative (red) correlation between metabolite concentration and expression level of selected gene data under drought conditions
Discussion
Contrasting drought tolerances of the two rice genotypes
The effectiveness of water uptake from a diverse soil environment is determined by root architecture. In drought condition due to increased soil resistance and decreased water availability, roots are unable to absorb or release water to the soil when the soil dries out. This leads to a decrease in osmotic potential and matric potential [33], as well as a decrease in turgor pressure and cell volume [34]. Root cells need to develop techniques to counteract water loss and its consequences. At times, the solution potential of the cells is reduced, increasing turgor pressure and allowing development to continue under water-deficient conditions [104].
Quantitative gene expression analysis
Consistent with our previous miRNA-seq and mRNA reports [31, 32], 13 genes involved in root growth and elongation and regulation of root primary metabolism were selected. Total RNA isolation was done using the TRIzol reagent (BioBasic-BS410A, Canada) as per the manufacturer’s guidelines. RNA quality was assessed on an agarose gel (1%). RNA quantity was found using a NanoPhotometer® spectrophotometer (NP80 NanoPhotometer, IMPLEN). The cDNA was reverse transcribed from the isolated RNA using the SuperScript First-Strand System for the RT-qPCR, which was performed using Invitrogen™ Kit. PerlPrimer v.1.1.21 software was used for primer designing from the transcribed region of the rice genes (sequences obtained from the RAP-DB database). qRT-PCR was accomplished using SYBR Green Master Mix (Eurogentec, Köln, Germany) in the ABI Prism 7900HT (Applied Biosystems, Foster City, CA) with the usual thermal cycling conditions (95 °C for 10 min, 95 °C for 15 s for 40 cycles, 60 °C for 1 min). The experiment was conducted in three biological replicates and two technical replicates were used. For the purpose of examining the dissociation curves for shoulders or extra peaks, the SDS 2.2.1 software (Applied Biosystems) was utilized. The expression values were normalized using the UBQ (Ubiquitin) gene as a housekee** gene [115]. LinRegPCR was used to calculate primer efficiency [116]. “Normalized expression of the genes of interest was calculated by dividing the average relative expression (primer efficiency P to the power of cycle number Ct) of the housekee** genes (H1) by the relative expression of the gene of interest (GOI): (GOI): ((PH1^CtH1)/2)/PGOI^CtGOI.”
Availability of data and materials
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Abbreviations
- FC:
-
Field capacity
- GC–MS:
-
Gas chromatography-mass spectrometry
- PCA:
-
Principal component analysis
- PCs:
-
Principal components
- PLSDA:
-
Partial least-squares-discriminant analysis
- VIP:
-
Variable importance in the projection
- QTL:
-
Quantitative trait locus
- RL:
-
Root length
- SA:
-
Surface area
- NIST:
-
National Institute of Standards and Technology
- GMD:
-
GOLM metabolome database
- SAM:
-
Significant Analysis of Metabolites
- cDNA:
-
Complementary DNA
- DAMs:
-
Differentially abundant metabolite
- OsTPP1:
-
Trehalose -phosphate phosphate
- SUT5:
-
SUCROSE TRANSPORTER 5
- DREB:
-
Dehydration-responsive element binding protein
- PRP5:
-
Proline-rich protein
References
Thoppurathu FJ, Ghorbanzadeh Z, Vala AK, Hamid R, Joshi M. Unravelling the treasure trove of drought-responsive genes in wild-type peanut through transcriptomics and physiological analyses of root. Funct Integr Genomics. 2022;22(2):1–19.
Dubey A, Kumar A. Abd_Allah EF, Hashem A, Khan ML: Growing more with less: breeding and develo** drought resilient soybean to improve food security. Ecol Ind. 2019;105:425–37.
Xu J, Pan C, Lin H, Ye H, Wang S, Lu T, Chen Q, Yang K, Lu M, Qian Q. A rice XANTHINE DEHYDROGENASE gene regulates leaf senescence and response to abiotic stresses. Crop J. 2022;10(2):310–22.
Muralikrishnan L, Padaria RN, Dass A, Choudhary AK, Kakade B, Shokralla S, El-Abedin TKZ, Almutairi KF, Elansary HO. Elucidating traditional rice varieties for consilient biotic and abiotic stress management under changing climate with landscape-level rice biodiversity. Land. 2021;10(10):1058.
Arif M, Jan T, Riaz M, Fahad S, Arif MS, Shakoor MB, Rasul F. Advances in rice research for abiotic stress tolerance: agronomic approaches to improve Rice production under abiotic stress. In: Advances in rice research for abiotic stress tolerance. Elsevier; 2019. p. 585–614.
Wu W, Ma BL, Whalen JK. Enhancing rapeseed tolerance to heat and drought stresses in a changing climate: perspectives for stress adaptation from root system architecture. Adv Agron. 2018;151:87–157.
Kang Z, Babar MA, Khan N, Guo J, Khan J, Islam S, Shrestha S, Shahi D. Comparative metabolomic profiling in the roots and leaves in contrasting genotypes reveals complex mechanisms involved in post-anthesis drought tolerance in wheat. PLoS ONE. 2019;14(3):e0213502.
Cabrera J, Conesa CM, Del Pozo JC. May the dark be with roots: A perspective on how root illumination may bias in vitro research on plant–environment interactions. New Phytologist. 2021;233(5):1988–97.
Prinsi B, Negri AS, Failla O, Scienza A, Espen L. Root proteomic and metabolic analyses reveal specific responses to drought stress in differently tolerant grapevine rootstocks. BMC Plant Biol. 2018;18(1):1–28.
Zeng W, Peng Y, Zhao X, Wu B, Chen F, Ren B, Zhuang Z, Gao Q, Ding Y. Comparative proteomics analysis of the seedling root response of drought-sensitive and drought-tolerant maize varieties to drought stress. Int J Mol Sci. 2019;20(11):2793.
Guo H, Ayalew H, Seethepalli A, Dhakal K, Griffiths M, Ma XF, York LM. Functional phenomics and genetics of the root economics space in winter wheat using high-throughput phenoty** of respiration and architecture. New Phytol. 2021;232(1):98–112.
Ghorbanzadeh Z, Hamid R, Jacob F, Asadi S, Salekdeh GH, Ghaffari MR. Non-coding RNA: Chief architects of drought-resilient roots. Rhizosphere. 2022;23:100572.
Maurel C, Nacry P. Root architecture and hydraulics converge for acclimation to changing water availability. Nature plants. 2020;6(7):744–9.
**ong R, Liu S, Considine MJ, Siddique KH, Lam HM, Chen Y. Root system architecture, physiological and transcriptional traits of soybean (Glycine max L.) in response to water deficit: a review. Physiologia Plantarum. 2021;172(2):405–18.
Mercado SAS, Bayona HAM. Evaluation of the cytotoxic potential of sodium hypochlorite using meristematic root cells of Lens culinaris Med. Sci Total Environ. 2020;701:134992.
Kim Y, Chung YS, Lee E, Tripathi P, Heo S, Kim K-H. Root response to drought stress in rice (Oryza sativa L.). Int J Mol Sci. 2020;21(4):1513.
Rejeth R, Manikanta CL, Beena R, Stephen R, Manju R, Viji M. Water stress mediated root trait dynamics and identification of microsatellite markers associated with root traits in rice (Oryza sativa L.). Physiol Mol Biol Plants. 2020;26(6):1225–36.
Song T, Das D, Yang F, Chen M, Tian Y, Cheng C, Sun C, Xu W, Zhang J. Genome-wide transcriptome analysis of roots in two rice varieties in response to alternate wetting and drying irrigation. Crop J. 2020;8(4):586–601.
Zhao Y, Zhou M, Xu K, Li J, Li S, Zhang S, Yang X. Integrated transcriptomics and metabolomics analyses provide insights into cold stress response in wheat. Crop J. 2019;7(6):857–66.
Fàbregas N, Fernie AR. The metabolic response to drought. J Exp Bot. 2019;70(4):1077–85.
Takahashi F, Kuromori T, Urano K, Yamaguchi-Shinozaki K, Shinozaki K. Drought stress responses and resistance in plants: from cellular responses to long-distance intercellular communication. Front Plant Sci. 2020;11:1407.
Guo X, **n Z, Yang T, Ma X, Zhang Y, Wang Z, Ren Y, Lin T. Metabolomics response for drought stress tolerance in chinese wheat genotypes (Triticum aestivum). Plants. 2020;9(4):520.
Wang Y, Zeng X, Xu Q, Mei X, Yuan H, Jiabu D, Sang Z, Nyima T. Metabolite profiling in two contrasting Tibetan hulless barley cultivars revealed the core salt-responsive metabolome and key salt-tolerance biomarkers. AoB Plants. 2019;11(2):plz021.
Ghaffari MR, Mirzaei M, Ghabooli M, Khatabi B, Wu Y, Zabet-Moghaddam M, Mohammadi-Nejad G, Haynes PA, Hajirezaei MR, Sepehri M. Root endophytic fungus Piriformospora indica improves drought stress adaptation in barley by metabolic and proteomic reprogramming. Environ Exp Bot. 2019;157:197–210.
Yue J, Wang L, Dou X, Wang Y, Wang H. Comparative metabolomic profiling in the roots of salt-tolerant and salt-intolerant maize cultivars treated with NaCl stress. Biol Plant. 2020;64:569–77.
Li C, Li L, Reynolds MP, Wang J, Chang X, Mao X, **g R. Recognizing the hidden half in wheat: root system attributes associated with drought tolerance. J Exp Bot. 2021;72(14):5117–33.
Guo Y, Huang G, Guo Q, Peng C, Liu Y, Zhang M, et al. Increase in root density induced by coronatine improves maize drought resistance in North China. Crop J. 2022;11(1):278–90.
Kirschner GK, Rosignoli S, Guo L, Vardanega I, Imani J, Altmüller J, Milner SG, Balzano R, Nagel KA, Pflugfelder D. ENHANCED GRAVITROPISM 2 encodes a STERILE ALPHA MOTIF–containing protein that controls root growth angle in barley and wheat. Proc Natl Acad Sci. 2021;118(35):e2101526118.
Kitomi Y, Itoh J-I, Uga Y. Genetic mechanisms involved in the formation of root system architecture. In: Rice genomics, genetics and breeding. Springer; 2018. p. 241–74.
Wang Y, Xu L, Shen H, Wang J, Liu W, Zhu X, Wang R, Sun X, Liu L. Metabolomic analysis with GC-MS to reveal potential metabolites and biological pathways involved in Pb & Cd stress response of radish roots. Sci Rep. 2015;5(1):1–13.
Abdirad S, Ghaffari MR, Majd A, Irian S, Soleymaniniya A, Daryani P, Koobaz P, Shobbar Z-S, Farsad LK, Yazdanpanah P. Genome-wide expression analysis of root tips in contrasting rice genotypes revealed novel candidate genes for water stress adaptation. Front Plant Sci. 2022;13:792079.
Ghorbanzadeh Z, Hamid R, Jacob F, Mirzaei M, Zeinalabedini M, Abdirad S, et al. MicroRNA Profiling of Root Meristematic Zone in Contrasting Genotypes Reveals Novel Insight into in Rice Response to Water Deficiency. J Plant Growth Regul. 2022:1–21.
Scharwies JD, Dinneny JR. Water transport, perception, and response in plants. J Plant Res. 2019;132(3):311–24.
Schimel JP. Life in dry soils: effects of drought on soil microbial communities and processes. Annu Rev Ecol Evol Syst. 2018;49:409–32.
Gupta A, Rico-Medina A, Caño-Delgado AI. The physiology of plant responses to drought. Science. 2020;368(6488):266–9.
Kapoor D, Bhardwaj S, Landi M, Sharma A, Ramakrishnan M, Sharma A. The impact of drought in plant metabolism: how to exploit tolerance mechanisms to increase crop production. Appl Sci. 2020;10(16):5692.
Fromm H. Root plasticity in the pursuit of water. Plants. 2019;8(7):236.
Sallam A, Alqudah AM, Dawood MF, Baenziger PS, Börner A. Drought stress tolerance in wheat and barley: advances in physiology, breeding and genetics research. Int J Mol Sci. 2019;20(13):3137.
Liu TY, Ye N, Song T, Cao Y, Gao B, Zhang D, Zhu F, Chen M, Zhang Y, Xu W. Rhizosheath formation and involvement in foxtail millet (Setaria italica) root growth under drought stress. J Integr Plant Biol. 2019;61(4):449–62.
Liu W, Wang J, Wang C, Ma G, Wei Q, Lu H, **e Y, Ma D, Kang G. Root growth, water and nitrogen use efficiencies in winter wheat under different irrigation and nitrogen regimes in North China Plain. Front Plant Sci. 2018;9:1798.
**ao S, Liu L, Zhang Y, Sun H, Zhang K, Bai Z, Dong H, Li C. Fine root and root hair morphology of cotton under drought stress revealed with RhizoPot. J Agron Crop Sci. 2020;206(6):679–93.
Karthika K, Rashmi I, Parvathi M. Biological functions, uptake and transport of essential nutrients in relation to plant growth. In: Plant nutrients and abiotic stress tolerance. Springer; 2018. p. 1–49.
Bouda M, Brodersen C, Saiers J. Whole root system water conductance responds to both axial and radial traits and network topology over natural range of trait variation. J Theor Biol. 2018;456:49–61.
Li Z, Zhang X, Zhao Y, Li Y, Zhang G, Peng Z, Zhang J. Enhancing auxin accumulation in maize root tips improves root growth and dwarfs plant height. Plant Biotechnol J. 2018;16(1):86–99.
Ciereszko I. Regulatory roles of sugars in plant growth and development. Acta Soc Botanicorum Pol. 2018;87(2):3583.
Saia S, Fragasso M, De Vita P, Beleggia R. Metabolomics provides valuable insight for the study of durum wheat: a review. J Agric Food Chem. 2019;67(11):3069–85.
Chen Y, Li C, Yi J, Yang Y, Lei C, Gong M. Transcriptome response to drought, rehydration and re-dehydration in potato. Int J Mol Sci. 2019;21(1):159.
Hong Y, Ni S-J, Zhang G-P. Transcriptome and metabolome analysis reveals regulatory networks and key genes controlling barley malting quality in responses to drought stress. Plant Physiol Biochem. 2020;152:1–11.
Thalmann M, Santelia D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017;214(3):943–51.
Saddhe AA, Manuka R, Penna S. Plant sugars: Homeostasis and transport under abiotic stress in plants. Physiol Plant. 2021;171(4):739–55.
Karakas B, Ozias-Akins P, Stushnoff C, Suefferheld M, Rieger M. Salinity and drought tolerance of mannitol-accumulating transgenic tobacco. Plant, Cell Environ. 1997;20(5):609–16.
Abebe T, Guenzi AC, Martin B, Cushman JC. Tolerance of mannitol-accumulating transgenic wheat to water stress and salinity. Plant Physiol. 2003;131(4):1748–55.
Du C, Li H, Liu C, Fan H. Understanding of the postgerminative development response to salinity and drought stresses in cucumber seeds by integrated proteomics and transcriptomics analysis. J Proteomics. 2021;232: 104062.
Michailidis M, Karagiannis E, Tanou G, Karamanoli K, Lazaridou A, Matsi T, Molassiotis A. Metabolomic and physico-chemical approach unravel dynamic regulation of calcium in sweet cherry fruit physiology. Plant Physiol Biochem. 2017;116:68–79.
Ogbaga CC, Stepien P, Johnson GN. Sorghum (Sorghum bicolor) varieties adopt strongly contrasting strategies in response to drought. Physiol Plant. 2014;152(2):389–401.
Rabbani N, Al-Motawa M, Thornalley PJ. Protein glycation in plants—An under-researched field with much still to discover. Int J Mol Sci. 2020;21(11):3942.
Chowdhury MK, Hasan M, Bahadur M, Islam M, Hakim M, Iqbal MA, Javed T, Raza A, Shabbir R, Sorour S. Evaluation of drought tolerance of some wheat (Triticum aestivum L.) genotypes through phenology, growth, and physiological indices. Agronomy. 2021;11(9):1792.
Jiang D, Chen W, Gao J, Yang F, Zhuang C. Overexpression of the trehalose-6-phosphate phosphatase OsTPP3 increases drought tolerance in rice. Plant Biotechnol Rep. 2019;13(3):285–92.
Joshi R, Sahoo KK, Singh AK, Anwar K, Pundir P, Gautam RK, Krishnamurthy S, Sopory S, Pareek A, Singla-Pareek SL. Enhancing trehalose biosynthesis improves yield potential in marker-free transgenic rice under drought, saline, and sodic conditions. J Exp Bot. 2020;71(2):653–68.
Deyanira Q-M, Estrada-Luna AA, Altamirano-Hernández J, Peña-Cabriales JJ, de Oca-Luna RM, Cabrera-Ponce JL. Use of trehalose metabolism as a biochemical marker in rice breeding. Mol Breeding. 2012;30(1):469–77.
Meena M, Divyanshu K, Kumar S, Swapnil P, Zehra A, Shukla V, Yadav M, Upadhyay R. Regulation of L-proline biosynthesis, signal transduction, transport, accumulation and its vital role in plants during variable environmental conditions. Heliyon. 2019;5(12):e02952.
Rai V. Role of amino acids in plant responses to stresses. Biol Plant. 2002;45(4):481–7.
You J, Zhang Y, Liu A, Li D, Wang X, Dossa K, Zhou R, Yu J, Zhang Y, Wang L. Transcriptomic and metabolomic profiling of drought-tolerant and susceptible sesame genotypes in response to drought stress. BMC Plant Biol. 2019;19(1):1–16.
Rao S, Qayyum A, Razzaq A, Ahmad M, Mahmood I, Sher A. Role of foliar application of salicylic acid and l-tryptophan in drought tolerance of maize. J Anim Plant Sci. 2012;22(3):768–72.
Zhao D, Zhang X, Wang R, Liu D, Sun J, Tao J. Herbaceous peony tryptophan decarboxylase confers drought and salt stresses tolerance. Environ Exp Bot. 2019;162:345–56.
Bowne JB, Erwin TA, Juttner J, Schnurbusch T, Langridge P, Bacic A, Roessner U. Drought responses of leaf tissues from wheat cultivars of differing drought tolerance at the metabolite level. Mol Plant. 2012;5(2):418–29.
Ackah M, Shi Y, Wu M, Wang L, Guo P, Guo L, ** X, Li S, Zhang Q, Qiu C. Metabolomics response to drought stress in morus alba L. variety Yu-711. Plants. 2021;10(8):1636.
Podlešáková K, Ugena L, Spíchal L, Doležal K, De Diego N. Phytohormones and polyamines regulate plant stress responses by altering GABA pathway. New Biotechnol. 2019;48:53–65.
Renault H, El Amrani A, Palanivelu R, Updegraff EP, Yu A, Renou J-P, Preuss D, Bouchereau A, Deleu C. GABA accumulation causes cell elongation defects and a decrease in expression of genes encoding secreted and cell wall-related proteins in Arabidopsis thaliana. Plant Cell Physiol. 2011;52(5):894–908.
Shelden MC, Dias DA, Jayasinghe NS, Bacic A, Roessner U. Root spatial metabolite profiling of two genotypes of barley (Hordeum vulgare L.) reveals differences in response to short-term salt stress. J Exp Bot. 2016;67(12):3731–45.
Chen CT, Kao CH. Osmotic stress and water stress have opposite effects on putrescine and proline production in excised rice leaves. Plant Growth Regul. 1993;13(2):197–202.
Parvathi M, Nataraja KN, Yashoda B, Ramegowda H, Mamrutha H, Rama N. Expression analysis of stress responsive pathway genes linked to drought hardiness in an adapted crop, finger millet (Eleusine coracana). J Plant Biochem Biotechnol. 2013;22(2):193–201.
Fadoul HE, Martínez Rivas FJ, Neumann K, Balazadeh S, Fernie AR, Alseekh S. Comparative molecular and metabolic profiling of two contrasting wheat cultivars under drought stress. Int J Mol Sci. 2021;22(24):13287.
Kumar M, Patel MK, Kumar N, Bajpai AB, Siddique KH. Metabolomics and molecular approaches reveal drought stress tolerance in plants. Int J Mol Sci. 2021;22(17):9108.
Itam M, Mega R, Tadano S, Abdelrahman M, Matsunaga S, Yamasaki Y, Akashi K, Tsujimoto H. Metabolic and physiological responses to progressive drought stress in bread wheat. Sci Rep. 2020;10(1):1–14.
Dilawari R, Kaur N, Priyadarshi N, Kumar B, Abdelmotelb K, Lal SK, Singh B, Tripathi A, Aggarwal S, Jat BS. Genome editing: a tool from the vault of science for engineering climate-resilient cereals. In: Harsh environment and plant resilience. Springer; 2021. p. 45–72.
Sharma A, Shahzad B, Rehman A, Bhardwaj R, Landi M, Zheng B. Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules. 2019;24(13):2452.
Naikoo MI, Dar MI, Raghib F, Jaleel H, Ahmad B, Raina A, et al. Role and regulation of plants phenolics in abiotic stress tolerance: An overview. Plant Signaling Mol. 2019;Chapter 9:157–68.
GorginiShabankareh H, Khorasaninejad S, Soltanloo H, Shariati V. Physiological response and secondary metabolites of three lavender genotypes under water deficit. Sci Rep. 2021;11(1):1–22.
Tiwari P, Srivastava D, Chauhan AS, Indoliya Y, Singh PK, Tiwari S, Fatima T, Mishra SK, Dwivedi S, Agarwal L. Root system architecture, physiological analysis and dynamic transcriptomics unravel the drought-responsive traits in rice genotypes. Ecotoxicol Environ Saf. 2021;207:111252.
Šamec D, Karalija E, Šola I, Vujcic Bok V, Salopek-Sondi B: The role of polyphenols in abiotic stress response: the influence of molecular structure. Plants 2021,10,118. In.: s Note: MDPI stays neu-tral with regard to jurisdictional clai-ms in … ; 2021.
Levi A, Paterson AH, Cakmak I, Saranga Y. Metabolite and mineral analyses of cotton near-isogenic lines introgressed with QTLs for productivity and drought-related traits. Physiol Plant. 2011;141(3):265–75.
Khan N, Ali S, Zandi P, Mehmood A, Ullah S, Ikram M, Ismail MAS, Babar M. Role of sugars, amino acids and organic acids in improving plant abiotic stress tolerance. Pak J Bot. 2020;52(2):355–63.
Huang L-b, Peng L-n, Yan X-h. Multi-omics responses of red algae Pyropia haitanensis to intertidal desiccation during low tides. Algal Res. 2021;58:102376.
Wendisch VF. Metabolic engineering advances and prospects for amino acid production. Metab Eng. 2020;58:17–34.
Munns R, Day DA, Fricke W, Watt M, Arsova B, Barkla BJ, Bose J, Byrt CS, Chen ZH, Foster KJ. Energy costs of salt tolerance in crop plants. New Phytol. 2020;225(3):1072–90.
LI P-c, YANG X-y, WANG H-m, Ting P, YANG J-y, WANG Y-y, et al. Metabolic responses to combined water deficit and salt stress in maize primary roots. J Integr Agric. 2021;20(1):109–19.
De Carvalho CC, Caramujo MJ. The various roles of fatty acids. Molecules. 2018;23(10):2583.
Lai S-H, Chye M-L. Plant acyl-CoA-binding proteins—their lipid and protein interactors in abiotic and biotic stresses. Cells. 2021;10(5):1064.
Hou Q, Ufer G, Bartels D. Lipid signalling in plant responses to abiotic stress. Plant, Cell Environ. 2016;39(5):1029–48.
Gundaraniya SA, Ambalam PS, Tomar RS. Metabolomic profiling of drought-tolerant and susceptible peanut (Arachis hypogaea L.) genotypes in response to drought stress. ACS omega. 2020;5(48):31209–19.
Zhang A, Yang X, Lu J, Song F, Sun J, Wang C, Lian J, Zhao L, Zhao B. OsIAA20, an Aux/IAA protein, mediates abiotic stress tolerance in rice through an ABA pathway. Plant Sci. 2021;308:110903.
Jogawat A, Yadav B, Lakra N, Singh AK, Narayan OP. Crosstalk between phytohormones and secondary metabolites in the drought stress tolerance of crop plants: a review. Physiol Plant. 2021;172(2):1106–32.
Gomes G, Scortecci K. Auxin and its role in plant development: structure, signalling, regulation and response mechanisms. Plant Biol. 2021;23(6):894–904.
Casanova-Sáez R, Mateo-Bonmatí E, Ljung K. Auxin metabolism in plants. Cold Spring Harb Perspect Biol. 2021;13(3):a039867.
Ahmed A, Tariq A, Habib S. Interactive biology of auxins and phenolics in plant environment. In: Plant phenolics in sustainable agriculture. Springer; 2020. p. 117–33.
Kord H, Fakheri B, Ghabooli M, Solouki M, Emamjomeh A, Khatabi B, Sepehri M, Salekdeh GH, Ghaffari MR. Salinity-associated microRNAs and their potential roles in mediating salt tolerance in rice colonized by the endophytic root fungus Piriformospora indica. Funct Integr Genomics. 2019;19(4):659–72.
Puértolas J, Larsen EK, Davies WJ, Dodd IC. Applying ‘drought’to potted plants by maintaining suboptimal soil moisture improves plant water relations. J Exp Bot. 2017;68(9):2413–24.
Pedrini S, Dixon KW. International principles and standards for native seeds in ecological restoration. Restor Ecol. 2020;28:S286–303.
Naseri NK, Sundararajan EA, Ayob M, Jula A. Smart Root Search (SRS): a novel nature-inspired search algorithm. Symmetry. 2020;12(12):2025.
Narisetti N, Henke M, Seiler C, Shi R, Junker A, Altmann T, Gladilin E. Semi-automated root image analysis (saRIA). Sci Rep. 2019;9(1):1–10.
Guo Q, Li X, Niu L, Jameson PE, Zhou W. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021;186(1):677–95.
Deng M, Zhang X, Luo J, Liu H, Wen W, Luo H, Yan J, **ao Y. Metabolomics analysis reveals differences in evolution between maize and rice. Plant J. 2020;103(5):1710–22.
Ghaffari MR, Shahinnia F, Usadel B, Junker B, Schreiber F, Sreenivasulu N, Hajirezaei MR. The metabolic signature of biomass formation in barley. Plant Cell Physiol. 2016;57(9):1943–60.
Pirttilä K, Balgoma D, Rainer J, Pettersson C, Hedeland M, Brunius C. Comprehensive Peak Characterization (CPC) in Untargeted LC–MS Analysis. Metabolites. 2022;12(2):137.
Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, **a J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic acids research. 2018;46(W1):W486–94.
Sumner L, Amberg A, Barrett D, Beale M, Beger R, Daykin C, et al. Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI). Metabolomics. 2007;(3):211–21.
Kopka J, Schauer N, Krueger S, Birkemeyer C, Usadel B, Bergmüller E, et al. GMD@ CSB. DB: the Golm metabolome database. Bioinformatics. 2005;21(8):1635–38.
Gerasimoska T, Ljoncheva M, Simjanoska M. MSL-ST: development of mass spectral library search tool to enhance compound identification. Bioinformatics. 2021;2021:195–203.
Chan ECY, Pasikanti KK, Nicholson JK. Global urinary metabolic profiling procedures using gas chromatography–mass spectrometry. Nat Protoc. 2011;6(10):1483–99.
Auler PA, Souza GM, da Silva Engela MRG, do Amaral MN, Rossatto T, da Silva MGZ, Furlan CM, Maserti B, Braga EJB. Stress memory of physiological, biochemical and metabolomic responses in two different rice genotypes under drought stress: the scale matters. Plant Sci. 2021;311:110994.
Khan N, Bano A, Rahman MA, Rathinasabapathi B, Babar MA. UPLC-HRMS-based untargeted metabolic profiling reveals changes in chickpea (Cicer arietinum) metabolome following long-term drought stress. Plant Cell Environ. 2019;42(1):115–32.
Pang Z, Chong J, Zhou G, de Lima Morais DA, Chang L, Barrette M, et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic acids research. 2021;49(W1):W388–96.
Ciasca B, Lanubile A, Marocco A, Pascale M, Logrieco AF, Lattanzio VM. Application of an integrated and open source workflow for LC-HRMS plant metabolomics studies. case-control study: metabolic changes of maize in response to Fusarium verticillioides infection. Front Plant Sci. 2020;11:664.
Rathod V, Hamid R, Tomar RS, Padhiyar S, Kheni J, Thirumalaisamy P, Munshi NS. Peanut (Arachis hypogaea) transcriptome revealed the molecular interactions of the defense mechanism in response to early leaf spot fungi (Cercospora arachidicola). Plant Gene. 2020;23:100243.
Kongprajug A, Chyerochana N, Mongkolsuk S, Sirikanchana K. Effect of quantitative polymerase chain reaction data analysis using sample amplification efficiency on microbial source tracking assay performance and source attribution. Environ Sci Technol. 2020;54(13):8232–44.
Acknowledgements
The authors thank the Agricultural Biotechnology Research Institute of Iran (ABRII) for providing lab facilities for this work.
Funding
This research was not supported by any agency.
Author information
Authors and Affiliations
Contributions
ZGH collected samples, performed laboratory procedures, prepared the first draft of the manuscript, and performed data analysis and figure preparation. RH assisted with data analysis, and figure preparation, and edited the manuscript. FJ and MZ assisted in editing the manuscript. GHS edited the manuscript and provided supervision. MRG conceptualized the experiments, data analysis, editing of the manuscript, and supervision. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental studies on plants were complied with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Identification results of differently accumulated metabolites between two rice genotypes under drought. Table S2. List of significant metabolites (between normal and drought conditions) identified through ANOVA with their p-value in root tip IR64. Table S3. List of significant metabolites (between normal and drought conditions) identified through ANOVA with their p-value in root tip Azucena. Table S4. The KEGG pathways of the altered metabolites exposure to drought stress in Azucena root samples. Table S5. The KEGG pathways of the altered metabolites exposure to drought stress in IR64 root samples. Table S6. List of primers with details of sequence and expression profile used for RT-qPCR validation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ghorbanzadeh, Z., Hamid, R., Jacob, F. et al. Comparative metabolomics of root-tips reveals distinct metabolic pathways conferring drought tolerance in contrasting genotypes of rice. BMC Genomics 24, 152 (2023). https://doi.org/10.1186/s12864-023-09246-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09246-z