
Abstract
Electrochemical water splitting presents an optimal approach for generating hydrogen (H2), a highly promising alternative energy source. Nevertheless, the slow kinetics of the electrochemical oxygen evolution reaction (OER) and the exorbitant cost, limited availability, and susceptibility to oxidation of noble metal-based electrocatalysts have compelled scientists to investigate cost-effective and efficient electrocatalysts. Bimetallic nanostructured materials have been demonstrated to exhibit improved catalytic performances for the oxygen evolution reaction (OER). Herein, we report carbon aerogel (CA) decorated with different molar ratios of Fe and Ni with enhanced OER activity. Microwave irradiation was involved as a novel strategy during the synthesis process. Inductively coupled plasma mass spectrometry (ICP-MS), X-ray diffraction (XRD), X-ray Photoelectron Spectroscopy (XPS), Scanning Electron Microscope (SEM), Energy dispersive X-ray spectroscopy (EDAX spectra and EDAX map**), Transmission Electron Microscope (TEM), High-Resolution Transmission Electron Microscope (HR-TEM), and Selected Area Electron Diffraction (SAED) were used for physical characterizations of as-prepared material. Electrochemical potential towards OER was examined through cyclic voltammetry (CV), chronoamperometry, and electrochemical impedance spectroscopy (EIS). The FeNi/CA with optimized molar ratios exhibits low overpotential 377 mV at 10 mAcm−2, smaller Tafel slope (94.5 mV dec−1), and high turnover frequency (1.09 s−1 at 300 mV). Other electrocatalytic parameters were also calculated and compared with previously reported OER catalysts. Additionally, chronoamperometric studies confirmed excellent electrochemical stability, as the OER activity shows minimal change even after a stability test lasting 3600 s. Moreover, the bimetallic (Fe and Ni) carbon aerogel exhibits faster catalytic kinetics and higher conductivity than the monometallic (Fe), which was observed through EIS investigation. This research opens up possibilities for utilizing bi- or multi-metallic anchored carbon aerogel with high conductivities and exceptional electrocatalytic performances in electrochemical energy conversion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The increasing energy demand and environmental pollution caused by industrial development promote extensive research towards clean, environmentally benign, and renewable energy production and storage technologies [1,2,9,10,11,12]. OER is a four-electron transfer process-based anodic half-reaction of electrochemical water splitting that has sluggish kinetics and low energy deficiency [13,14,23,24]. Binary transition metal catalysts seem to be efficient for OER. Particularly Ni–Fe catalysts are shown to exhibit activity comparable to highly active noble metal-based catalysts and are appropriate for real-world applications [25].
Ullal et al. synthesized and created nanocrystalline Ni–Fe alloy coatings by electrodeposition as an effective, reliable, and affordable electrocatalytic material for HER and OER. They demonstrated that the composition, surface morphology, and phase structures of Ni–Fe alloy coatings had an impact on their particular catalytic characteristics [26]. Makaela et al. prepared NiFeOx oxyhydroxide electrocatalyst for OER in different mediums (alkaline to neutral). The prepared electrocatalysts showed excellent electrocatalytic activity in alkaline media with low overpotential (below 300 mV) in 0.1 M KOH [25]. Daoxin et al. prepared NiFe-layered double hydroxide (NiFe-LDH) hybrid catalysts through a hydrothermal method and studied its catalytic activity towards OER. With a low overpotential of 292 mV at 50 mAcm−2 current density, the material performed admirably. Excellent morphology, a large number of functional groups, and a synergistic effect are all responsible for this outstanding activity [27]. Yu et al. reported that the NiFe LDH@Cu nanoarray exhibits exceptional activity, requiring only 315 mV overpotential to deliver a huge current density of 1000 mAcm2, and 199 mV to deliver 10 mAcm2 for OER in 1.0 M KOH [28]. Beatrice Ricciardi et al. prepared Fe–Ni decorated carbon matrix as an efficient bifunctional electrocatalyst for OER and ORR. Prepared electrocatalyst showed excellent activity with high current density, low overpotential and small Tafel slop value (68.2 mA dec−1) [29]. Shuntian Huang et al. adopted sol–gel leading to a supercritical drying strategy for the synthesis of Ru-promoted Fe and Ni anchored N-doped carbon aerogel for HER/OER catalysis. Prepared three-dimensional electrocatalyst possessed a high surface area of 227.7 m2.g−1, low overpotential and high turnover frequency of 0.1107 s−1 [30]. Yiwen Zhang et al. reported NiFe-anchored GO as an efficient electrocatalyst of OER electrocatalysis. Prepared material exhibited a high surface area (242.4 m2g−1), low overpotential value, and a smaller Tafel slop of 42.0 mV dec−1 [31].
The specific surface area of the active metal, the catalyst's reducibility, and the dispersion of the active site can all be improved by using catalytic support materials. The 3D carbonaceous material group includes a family of organic aerogels called carbon aerogel (CA). High specific surface area, porosity, and low density are some of its exceptional surface characteristics. Due to its reticulated network, it possesses excellent mechanical strength and electrical conductivity. These characteristics make them appropriate support materials for metal catalysts [32]. Based on the above studies, Ni and Fe-supported carbon aerogel was selected in order the study their activity for electrochemical oxygen evolution reaction.
In this research, CA-supported Ni–Fe catalysts were developed for OER. CA was synthesized by sol–gel method, supercritical drying and pyrolysis processes. A novel microwave irradiation strategy was adopted for the homogeneous dispersion of Fe and Ni on carbon aerogel support material. In previous studies, similar electrocatalysts were prepared through sol–gel followed by supercritical drying [32, 33] and pyrolysis followed by freeze drying [34]. To ensure the porous structure of the CA catalyst support material, which is interconnected with each other and does not collapse within the well-designed 3D matrix, drying was done especially with supercritical carbon dioxide (CO2). With this morphology, it is aimed to obtain catalysts with extremely large electrochemically active surface areas. Then, Ni–Fe/CA catalysts using three different ratios of Ni and Fe metals were prepared by microwave irradiation method. Additionally, the Fe/CA catalyst was synthesized to compare the effects of Ni metal and loading rate on catalyst performance. In this way, both the different distributions of Ni and Fe transition metals on the CA support and the effects of these differences on catalyst performances were examined. The structural characteristics of the prepared catalysts have been examined using different physical characterization techniques, including Inductively Coupled Plasma-Mass Spectrometry (ICP-MS), X-Ray diffraction (XRD), X-ray Photoelectron Spectroscopy (XPS), Scanning Electron Microscope (SEM), Energy dispersive X-ray spectroscopy (EDAX spectra and EDAX map**), Transmission Electron Microscope (TEM), High Resolution Transmission Electron Microscope (HR-TEM), and Selected Area Electron Diffraction (SAED). To determine how metal loading of the CA aerogel support material affects OER performance, electrochemical investigation using cyclic voltammetry (CV), chronoamperometry, and electrochemical impedance spectroscopy (EIS) in alkaline media (1 M KOH: Methanol) was conducted.
2 Experimental
2.1 Materials
Resorcinol (99%), formaldehyde (w/w ≥ 34.5%), sodium carbonate (Na2CO3), and acetone (99.5%) used in carbon aerogel synthesis were obtained from Sigma Aldrich. High-purity carbon dioxide (CO2) and nitrogen (N2) gases used in various stages of synthesis were obtained from Habaş company. Nickel (II) chloride hexahydrate (NiCl2.6H2O) from LA CHE MA, Iron (III) chloride hexahydrate (FeCl3.6H2O, 98%) from Carlo Erba Reagent, potassium hydroxide (KOH) from Merck and ethylene glycol (extra pure) from ISOLAB Chemicals were used for the catalyst synthesis. Nafion solution (15%, Ion Power, Inc.) and 1,2-propanediol (C3H8O2, ≥ 99.5%, Sigma–Aldrich) were used for electrode solutions.
2.2 Synthesis of carbon aerogel (CA)
CA was synthesized by polymerization of resorcinol (R) and formaldehyde (F) in the presence of a catalyst (C, Na2CO3) and using the sol–gel method [35]. Sol solution prepared based on R/C = 200, R/W = 0.02, and R/F = 0.5 molar percent ratios was placed in a closed glass tube and incubated for 24 h at room temperature, 24 h at 50 °C, and 72 h at 90 °C. The gel in the glass tube, which was removed from the oven, was thrown into acetone in a beaker and left for 24 h. As a result of this process, the gel was placed in the supercritical reactor with acetone and extracted with supercritical CO2 at 2000 psi and 50 °C. The R-F aerogel obtained after drying with supercritical CO2 was pyrolyzed at 1000 0C for 4 h and in an N2 environment to reach the final CA structure.
2.3 Synthesis of Fe/CA and Ni–Fe/CA catalysts
The synthesis of metallic catalysts using CA support was carried out using the microwave irradiation method [36]. NiCl2.6H2O and FeCl3.6H2O were used as metal precursors and the loading process was carried out in one step. Support material and metal loading ratios were determined as CA-Ni–Fe:10-7-3, CA-Ni–Fe:10-5-5, and CA-Ni–Fe:10-3-7 which were named as Ni70Fe30/CA, Ni50Fe50/CA, and Ni30Fe70/CA, respectively. In addition, the Fe/CA catalyst was also synthesized to see the effect of Ni do** on the catalysts on the performance and for comparison. The nominal amount of Fe loaded in the catalyst was 30%. The predetermined amounts of CA, NiCl2.6H2O, FeCl3.6H2O and 20 ml of distilled water were mixed in a beaker. It was then dissolved in an ultrasonic bath for 30 min. 35 ml of ethylene glycol was added to this mixture and stirred for 3 h in a magnetic stirrer. 3 M KOH was used to increase the pH to 12. In this way, the adsorption of metal precursors to the CA surface was ensured. The mixture was heated in a microwave oven at 800 W for 1 min followed by flash cooling in an ice bath. It was washed in a centrifuge with acetone and distilled water until pH: 7-7.5. Finally, it was dried in an oven at 90–100 °C for 24 h.
3 Characterization
3.1 Physical characterizations
Inductively coupled plasma-mass spectrometer (ICP-MS, Agilent 7800) was used to determine the amount of metal loaded on the support material. X-Ray diffraction (XRD, PANalytical Empyrean, CuK∞, ʎ = 1.5406 Ǻ, scan range: 100 ≤ 2θ ≥ 900) device was used to obtain information about the crystal structures of the catalysts. X-ray Photoelectron Spectroscopy (XPS, Specs-Flex XPS, Energy range: 200 eV–4 keV) analysis was used to study and understand the surface chemistry and electronic structure of the catalysts. Scanning Electron Microscope (SEM) analysis was used to reveal the presence of metal nanoparticles in the catalysts and the morphology of the CA support surface in which they were incorporated. Energy dispersive X-ray spectroscopy (EDAX) and map** analyzes were used to characterize the elemental composition and distribution in the catalysts. Transmission Electron Microscope [TEM, Hitachi HighTech HT7700, Resolution values: 0.204 nm (100 kV)–0.144 nm (120 kV)] and High Resolution Transmission Electron Microscope (Jeol 2100F HR-TEM, Orius SC1000 Model 832 11 Megapixel CCD camera) was used to determine the simultaneous micro/nano and crystal structure of catalysts and support material. Selected Area Electron Diffraction (SAED) analyses were performed using the HR-TEM device. SAED analysis has been used to obtain crystallographic (crystal structure, orientation, and phase composition) information about catalysts.
3.2 Electrochemical characterizations
The potentiostat (Gamry 1010B) was used to conduct the electrochemical analysis of the as-synthesized materials. Ag/AgCl (3 M KCl), modified glassy carbon (3 mm), and Pt wire were utilized as the reference, working, and counter electrode, respectively, in a standard three-electrode cell assembly [37]. Electrode modification was done by the ink paste method. 1 M KOH was used as a supporting electrolyte and a small amount of methanol was also added to envision its effect on the oxygen evolution reaction. The electrochemical investigation was carried out by conducting cyclic voltammetry (CV), chronoamperometry, and electrochemical impedance spectroscopy (EIS). CV was performed at 100 mVs−1 scan rate, chronoamperometric analysis was carried out in 1 M KOH by application of 1.4 V potential for 3600 s. EIS measurements were performed with 5 mV AC and 1.1–1.5 V DC voltage and a variable frequency of 0.1 Hz to 20 kHz.
3.3 Electrode modification
Before electrochemical studies, the glassy carbon electrode surface was polished with alumina slurry and then modified with as-synthesized electrocatalyst material. Electrode inks were prepared by mixing pure water (1 ml), 1,2-propanediol (1 ml), Nafion solution (165 µl), and catalyst material. The mass loading ratio of the active catalyst material was determined as 1 mg/cm2. After the electrode ink was mixed very well in the magnetic stirrer, 3 μl was dropped on the GC electrode surface with the help of a micropipette. The electrode surface was left to dry at room temperature. This modified electrode was employed for electrochemical studies [38].
4 Results and discussion
4.1 Physical characterizations
Before synthesizing Ni–Fe/CA catalysts, CA was synthesized as catalyst support material. The BET surface area of CA was measured as 664 m2/g with an average pore diameter of 9.6 nm [35]. The high surface area and favorable pore structure of CA make it attractive to be used as a catalyst support material. TEM images of CA are given in Fig. 1. CA consists of structurally interconnected spherical particles. The particles form a network structure to provide highly developed mesopores [39]. The superior surface properties of CA make it a suitable substrate for the homogeneous dispersion of metal catalyst nanoparticles without agglomeration.

TEM images (500 nm and 200 nm) of CA
The catalytic activity of a catalyst is highly dependent on the proportion of metal charged to the support surface, the types of metal phases, and the size of the metal nanoparticles. To measure these parameters, ICP MS, XRD, XPS, SEM, EDAX, TEM, HR-TEM and SAED analyses of Ni–Fe/CA catalysts were performed, respectively. ICP-MS analysis was performed to determine the metal loading percentages of Fe/CA and Ni–Fe/CA catalysts synthesized by microwave irradiation method and the results are given in Table 1. The loaded metal ratio in the Fe/CA catalyst is 24.2%. In Ni–Fe/CA catalysts, on the other hand, the loaded total metal ratios vary between 36 and 40%. Ni ratio in Ni70Fe30/CA, Ni50Fe50/CA, and Ni30Fe70/CA catalysts decreased as intended, and Fe ratio increased as well. Due to these differences in metal loading ratios, the differences in the distribution of metals on the CA support surface vary significantly and the nanoparticle-crystal sizes were investigated by XRD and TEM analyses. In addition, the effect of structural differences of these Ni–Fe/CA catalysts on electrochemical performance was also investigated.
XRD analyses were performed to obtain information about the crystal purity and crystal structure of the catalysts. XRD spectra of synthesized catalysts are given in Fig. 2. All catalysts have a diffraction peak at 2θ = 23.50. This peak corresponds to the C(002) plane and indicates the presence of graphitic carbon [40]. It represents the CA used as the catalyst support material. In the spectrum of the Fe/CA catalyst, there are distinct diffraction peaks at 2θ = 35.50, 44.50, and 620 angles. These peaks correspond to the Fe3O4(311), Fe(110) and Fe(200) planes, respectively. The Fe(110) and Fe(200) planes represent characteristic body-centered cubic (bbc) iron (JCPDS card no. 87–0722) [41]. The Fe3O4(311) plane shows that some iron oxide other than elemental iron (Fe0) is formed during the reaction [42]. There are several distinct diffraction peaks in the spectra of Ni–Fe/CA catalysts. The angles of these peaks are 2θ = 11.50, 350, 400, 46.50, 500, 600, 61.50, 650, 74.50 and 800. The diffraction peaks at 2θ = 46.50, 650 and 800 angles correspond to the Fe(110), Fe(200), and Fe(211) planes. They are crystal structures of elemental iron. The diffraction peaks at 2θ = 400, 500 and 74.50 correspond to the Ni–Fe(111), Ni–Fe(200) and Ni–Fe(220) planes, respectively. These three characteristic peaks are explained by the face-centered cubic (fcc) Ni3Fe alloy phase (combination of α-Fe(Ni) phase (kamacite) and γ-Fe–Ni (taenite) phase (JCPDS card no. 38-0419) [43,44,45]. Taenite contains more Ni than kamacite. The increase in Ni content causes taenite to be in face-centered crystal form, while the high Fe content in kamacite causes it to be body-centered. This difference is due to the similar size of Ni and Fe, but different interatomic magnetic and quantum interactions [46].

XRD spectra of catalysts, a Fe/CA, b Ni30Fe70/CA, c Ni50Fe50/CA, d Ni70Fe30/CA
In addition, differences in the amount of metal precursors used in catalyst synthesis cause differences in the interactions between the support and the metal and between the metals themselves. As the Ni ratio in the catalysts increases, the peak intensities of the oxide and hydroxide-containing phase groups increase. The distinct diffraction peak at the angle of 2θ = 350 belongs to the Ni-Fe3O4(311) crystal structure [or NiFe2O4(311)-nickel ferrite phase] and indicates fcc nanoparticle formation with spinel phase [47, 48]. The diffraction peaks at 2θ = 11.50, 600 and 61.50 angles correspond to the Ni–Fe-OH(003), Ni–Fe-OH(110) and Ni–Fe-OH(113) planes. These crystal structures contain double hydroxide (JCPDS card no. 41–1390) [43, 49].
These phases (Ni-Fe3O4 and Ni–Fe-OH) can form in the surface layer of the catalyst due to the oxidizing reaction of nickel and iron during the drying process of the catalysts [50]. González-Castaño et al. synthesized NiFe/C catalysts by wet impregnation method using three different carbon supports (biochar: CA, CB, CC). Some low-density peaks of Ni0 phases were observed, possibly due to partial re-oxidation of Ni particles in contact with air on the surfaces of some catalysts. Therefore, certain differences in Ni0 crystal sizes were found. Ni0 crystallite sizes of 20 nm, 9 nm, and 10 nm were calculated for NiFe/CA, NiFe/CB, and NiFe/CC catalysts, respectively [51].
The Debye–Scherrer formula was used to calculate the size of the crystal forms in the catalysts [52]. All crystal sizes of distinct diffraction peaks were calculated. The maximum, minimum, and average crystal sizes are summarized in Table 1. The average crystal size in the Fe/CA catalyst is 5.81 nm. The average crystal sizes of Ni70Fe30/CA, Ni50Fe50/CA and Ni30Fe70/CA catalysts were calculated as 7.48 nm, 9.65 nm and 4.85 nm, respectively. These crystal sizes are quite good for bimetallic catalysts. This shows that the metal particles dispersed on the support surface are dispersed without agglomeration.
XPS analysis is an important technique for studying the surface chemistry (elemental composition of the catalyst surface and different oxidation states of Ni and Fe) of Ni–Fe/C catalysts that are prominent in electrocatalysis, especially OER. For example, the presence of Ni2+ or Fe3+ may be indicative of certain active sites that are more effective at catalyzing OER. Additionally, this technique is very widely used to understand the electronic structure (electron transfer and orbital interactions) of catalysts. XPS provides information on the electronic structure of the catalyst by analyzing the binding energies of core electrons (e.g., Ni 2p, Fe 2p). This information helps to understand how electrons are transferred during the catalytic process and how orbital interactions between metal centers and carbon support contribute to catalytic activity [53, 54].
The XPS general spectra of the catalysts are given in Fig. 3. For the Fe/CA catalyst, the regions with binding energies of 284.0 eV, 530.0 eV, and 710.0 eV correspond to C 1 s, O 1 s, and Fe 2p3/2 elements, respectively. For the Ni30Fe70/CA catalyst, the regions with binding energies of 282.0 eV, 530.0 eV, 713.0 eV, and 858.0 eV correspond to the elements C 1 s, O 1 s, Fe 2p3/2, and Ni 2p3/2, respectively. For the Ni50Fe70/CA catalyst, the regions with binding energies of 282.0 eV, 531.0 eV, 713.0 eV, and 859.0 eV correspond to the elements C 1 s, O 1 s, Fe 2p3/2, and Ni 2p3/2, respectively. For the Ni70Fe30/CA catalyst, the regions with binding energies of 283.0 eV, 530.0 eV, 713.0 eV, and 859.0 eV correspond to the elements C 1 s, O 1 s, Fe 2p3/2, and Ni 2p3/2, respectively. Atomic % ratios of elemental analysis data obtained from XPS general spectra are given in Table 2.

XPS general spectra of catalysts
Ni 2p, Fe 2p, C 1 s and O 1 s spectra of the catalysts are given in Fig. 4. The bonds and binding energies corresponding to the peaks in these spectra are indicated on the spectrum. Additionally, the % rates of the species belonging to the bonds are indicated on these spectra. There are distinct peaks in the binding energy range of 845–885 eV of the Ni 2p spectra of the catalysts. These peaks arise from bonds belonging to Ni 2p3/2 (Ni2+) and Ni 2p1/2 (Ni2+) types. These species represent Ni-OH bonds [55, 56]. There are distinct peaks in the binding energy range of 700–740 eV of the Fe 2p spectra of the catalysts. These peaks arise from bonds belonging to Fe 2p3/2 (Fe3+) and Fe 2p1/2 (Fe3+) species [57]. These species contribute to Fe-OH bonds [56]. There are distinct peaks in the binding energy range of 528–534 eV of the O 1 s spectra of the catalysts. These peaks arise from bonds belonging to lattice oxygen, hydroxyl oxygen and incidental carbon–oxygen species. That is, it is generally explained by physically and chemically absorbed molecular water [58]. There are distinct peaks in the binding energy range of 280–290 eV in the C 1 s spectra of the catalysts. These peaks originate from bonds belonging to metal-C, C = C, C–C and C–O types [61, 62]. SEM analyzes of the synthesized catalysts were performed and the images are give in Fig. 4. First, the SEM image at 500 µm is given. Then, EDAX map**s were given on the same SEM image. EDAX maps for all catalysts typically reveal the presence of primary components Ni, Fe, C and O. Except for the Fe/CA catalyst, it is seen that the Ni and Fe nanoparticles in the other catalysts are distributed homogeneously on the CA surface without agglomeration.


SEM images (500 µm) and EDAX map** images of catalysts a Fe/CA, b Ni30Fe70/CA, c Ni50Fe50/CA, d Ni70Fe30/CA
EDAX spectra are used to obtain information about the elemental composition of catalysts. EDAX spectra of the catalysts are given in Fig. 6 and the peaks belonging to the primary components (Ni, Fe, C, O) are clearly visible. Weight and atomic % values obtained in EDAX spectra are given in Table 3. The weight % values of the elements in the structure of the catalysts are approximately in agreement with the ICP-MS analysis data given in Table 1. When the atomic % values of the elements are compared with the XPS analysis data given in Table 2, deviations from these values are observed. Because only certain chemical species (e.g., Ni 2p3/2, Fe 2p3/2) were considered in the general XPS analysis data. Other species are extensively explained by the XPS spectra given in Fig. 4. For example, while the EDAX atomic % of the Fe element in the Fe/CA catalyst is 5.2, the value of the Fe 2p3/2 type is given as 1.9% in the general XPS data table. This situation is clearly seen in the Fe 2p spectrum given in Fig. 4a. According to this spectrum, 27.3% of the Fe 2p chemical species is Fe 2p3/2, while 72.7% is Fe 2p1/2. When the necessary proportion calculation was made based on the Fe 2p spectrum, the XPS atomic % ratio of Fe was calculated as 5.1. This value agrees with the EDAX atomic % ratio of the element Fe.

EDAX spectra of catalysts
TEM and HR-TEM analyzes were performed to provide detailed images of the structure of Ni–Fe/C catalysts at the nanoscale and to examine the interaction between Ni–Fe nanoparticles and carbon support. Images are given in Fig. 7. In the TEM images of the catalysts, it is seen that Fe/CA and Ni70Fe30/CA catalysts are mostly nanospheres, in a mixed-shaped structure consisting of nanoshells and nanocubes. In Ni50Fe50/CA and Ni30Fe70/CA catalysts, the distribution of spherical nanoparticles is generally dominant. It is seen that the Ni–Fe catalyst nanoparticles are generally homogeneously distributed on the CA support surface, although they are locally agglomerated. Zhang et al. synthesized the unsupported Fe–Ni catalyst by the reduction-substitution method. Due to the lack of support and interaction at the nanoscale, it was observed that the aggregation of the prepared catalyst particles occurred easily [63]. CAs have been accepted as ideal support materials due to their superior surface properties (porosity, ideal pore size distribution, low density, etc.). TEM and HR-TEM images of Ni–Fe/CA catalysts show that metal nanoparticles have dark and bright areas. In the literature, it is estimated that the dark black area is kamacite without phase transition and the bright area is the formed γ-Fe–Ni taenite phase [47]. During the catalyst synthesis, the synthesis method, the type of precursor salts, reactant concentrations, pH, the properties of the support material used, and the presence of the support material are the parameters that affect the structure of the final catalyst. The size, shape, and distribution of metal nanoparticles are the most important parameters affecting catalyst performance. For example, the shape of the crystal changes the catalyst surface properties and therefore the atomic arrangements at each surface, which has a profound effect on the various properties of the crystal. Sayed et al. synthesized iron oxide in different shapes (nanorod, nanoshell, degraded cubes, nanocubes, porous spheres, and self-oriented flowers) using the same synthetic method and different Fe precursor salts [64].


TEM (left column) and HR-TEM (right column) images of catalysts a Fe/CA, b Ni30Fe70/CA, c Ni50Fe50/CA, d Ni70Fe30/CA, and d SAED analysis of Ni70Fe30/CA
SAED analysis was performed for the Ni70Fe30/CA catalyst to help identify the different crystalline phases present. The diffraction pattern of the Ni70Fe30/CA catalyst are given in Fig. 4d. With this analysis, various crystal forms such as Ni, Fe and their alloys or oxides that may be present in the catalyst can also be identified. A few circular lines are clearly visible in the diffraction pattern of the catalyst. These circular lines correspond to the (111), (200), (220), (311), (110) and (113) crystal planes, respectively, from the inner ring to the outer ring. These results are also in agreement with the XRD spectrum of the Ni70Fe30/CA catalyst [65, 66].
4.2 Electrochemical characterization
To investigate the electrochemical behavior of Fe/CA, Ni70Fe30/CA, Ni50Fe50/CA, and Ni30Fe70/CA in 1 M KOH followed by the addition of methanol at a scan rate of 100 mVs−1, linear sweep voltammetry was performed. Comparative cyclic voltammograms of each catalyst in 1 M KOH solution are shown in Fig. 8a. Fe/CA showed minimum peak current among all catalysts, which confirmed that the inclusion of Ni in the catalyst enhanced the OER activity. Congling Hu and co-workers [67] proposed the universal OER mechanism in which the M-OH bond is formed by the oxidation of OH ion adsorbed on the catalyst surface, while the second step involves the formation of metal oxide (M–O) by proton-electron transfer. Here, two M–O species convert either directly to oxygen while leaving the active sites of the catalyst or transform into M–OOH and then release oxygen after another proton-electron coupled reaction.

Comparative linear sweep voltammograms of prepared electrocatalyst in a 1 M KOH and b 1 M KOH + 1 M Methanol, c comparison of I-V response in bare GC (blue), 1 M KOH (red), 1 M KOH + 1 M Methanol (black) of Ni30Fe70/CA modified electrode, and d voltammogram indicating onset potential for Ni30Fe70/CA modified electrode
In this regard, OER responses were recorded in KOH electrolyte in the presence of methanol. The OER activity was enhanced multi-fold, and CV profiles are presented in Fig. 8b, c, rendering methanol a strong facilitator for OER. The formation of salt-like species (MeO-K +) decreases the diffusion of OH- ions towards the electrode–electrolyte interface and increases their adsorption of the catalyst surface, hence improving the kinetics of the water oxidation process [68,69,70,71,72,73,74]. Voltammetry measurements reveal that methanol significantly enhances the small current associated with OER in KOH, leading to a large peak current shown in Fig. 4c. Onset potential is an important parameter for the determination of the efficiency of a catalyst [75]. It is the potential that each electrochemical process starts and increases in output current observed in a cyclic voltammogram. Ni30Fe70/CA showed the lowest onset potential which is represented in Fig. 8d. This observation serves as strong evidence for the effectiveness of methanol as a facilitator for OER and confirms the superior electrocatalytic performance of the synthesized materials.
4.2.1 Calculation of kinetic parameters
The mechanism of the oxygen evolution reaction was studied through kinetic analysis, which involves a variation of several parameters like scan rate and methanol concentration. To investigate the diffusion-controlled mechanism of OER, the scan rate was increased from 20 to 100 mVs−1. The results revealed that the process is diffusion-controlled since the peak current increased with the scan rate (Fig. 9a). From the Randles–Sevcik equation, the experimental diffusion coefficient was derived [76, 77].
where Ip is anodic peak current (A), n is a total number of electrons (4 for OER), α is transfer coefficient, nα is number of electrons in rate determining step (nα = 1), A is active surface area (0.07cm2), C is concentration of electrolyte (mol.cm−3), D° is diffusion coefficient (cm2.s−1), and ν is scan rate (V.s−1) respectively.

a Cyclic voltammograms in 1 M KOH + 1 M Methanol at scan rate of 20–100 mVs−1 for Ni70Fe30/CA modified electrode, b Plot between anodic peak current and the square root of scan rate for OER at all Carbon aerogel supported modified GCE, c Cyclic voltammograms in different Methanol concentrations (0–2 M) at 100 mVs−1 scan rate for Ni30Fe70/CA modified electrode, and d Plot between anodic peak current and methanol concentration for OER at all Carbon aerogel supported modified GCE
The slope of the curve between the peak current and the square root of the scan rate exhibited in Fig. 9b was used to compute the diffusion coefficient. the following equation was used to determine the mass transfer coefficient [76, 77]:
where, mT is mass transport coefficient (cm.s−1), D° is diffusion coefficient (cm2.s−1), R is gas constant (J k−1 mol−1), T is temperature (K), F is Faraday’s constant (96,500 C.mol−1) and ν is scan rate (V.s−1).
Transfer coefficient (α) was calculated from the Eq. 3 in which Ep is peak potential, Ep/2 is potential when Ip = Ip/2, α is transfer coefficient and n is total number of electrons [77].
Onset potential was recorded with respect to Ag/AgCl reference electrode and then converted to RHE by using the following equation [37].
Onset potential was observed for all prepared electrocatalysts which is in the range of 1.501 to 1.543 V in 1 M KOH + 1 M methanol at 100 mVs−1 sweep rate.
Retrieved electrochemical parameters are summarized in Table 4.
From Table 4, the compositions with Fe-70% showed excellent activity toward oxygen evolution reaction. Low onset potential for this catalyst is witnessed for better performance.
The concentration of methanol was increased from 0.25 M to 2 M, and the LSV response was assessed for all materials. Figure 9c illustrates the effect of changing methanol concentrations on the OER using a CA-supported NiFe-modified electrode. An explicit trend of increasing current density with methanol concentration is demonstrated. We can see from the electrochemical response that methanol speeds up the OER process and raises the output current. We did not increase KOH concentration because it decreases the stability of the reference electrode. Primarily, a sharp rise in OER current was observed that was evident to the optimal composition (Ni30Fe70/CA). Upon increasing methanol concentration, the formation of MeO-K + (hence more hydroxyl ions form) increases, which facilitates the rate of the OER process. However, at higher methanol concentrations because of the formation of the complex-like molecule catalyst active sites become less available for hydroxyl ions hence there is a decrease in OER rate and output current [77, 78]. The data acquired from plots between the peak current and methanol concentration were also used to compute the heterogeneous rate constant. Calculation of the heterogeneous rate constant was done by using the Reinmuth equation [77, 78].
In this equation, Ip is peak current (mA), n is the total number of electrons, F is Faraday’s constant (96,500 C.mol−1), A is the active surface area of the electrode (0.07 cm−2), C is the concentration of methanol, and k° is the heterogeneous rate constant.
The plot of Ip vs. methanol concentration for different modified electrodes is presented in Fig. 9d. All the calculated electrocatalytic parameters are summarized in Table 5.
From the results, it can be concluded that Ni30Fe70/CA electrocatalyst showed excellent performance among all, with a larger diffusion coefficient, mass transport coefficient, and heterogeneous rate constant.
4.2.2 Determination of active surface area
To calculate the active surface area, cyclic voltammetry was used in a solution of 5 mM Potassium ferricyanide and 1 M potassium chloride at a scan rate of 100 mVs−1 and a potential window of −0.2 to 0.6 V. Reversible peaks for the Fe2+/Fe3+ redox pair were recorded on the cyclic voltammogram (Fig. 10). Peak current was noted, and the active surface area was computed using the equation below.
where Ipa is anodic peak current (A), n is total number of electrons (1), A is active surface area, C is concentration of electrolyte (mol.cm−3), D° is diffusion coefficient (0.76 × 10–6 cm2.s−1), and ν is scan rate (V.s−1), respectively.

Cyclic voltammograms of electrocatalyst-modified electrodes recorded in 5 mM K4[Fe(CN)6] + 1 M KCl at 100 mVs−1
4.2.3 Stability test
High activity and long-lasting stability are crucial for catalysts to be useful in real-world applications [79]. The synthesized material needs to be both mechanically and chemically stable in the environment in which it will be used. Chronoamperometry is a sophisticated electrochemical technique used to assess the stability of electrocatalyst material [80].
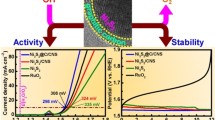
Chronoamperometric responses for all prepared electrocatalysts were recorded in 1 M KOH electrolyte using a potential of 1.4 V for 3600 s. The I-V response demonstrated that the current densities remained stable throughout the chronoamperometric scans, as shown in Fig. 11a.

a Chronoamperometric stability test for all electrocatalysts in 1 M KOH for 3600 s, b Tafel slopes of all prepared electrocatalysts in 1 M KOH electrolyte, c Tafel slopes of all prepared electrocatalysts in 1 M KOH + 1 M Methanol, d overpotential at various current densities in 1 M KOH (with and without methanol), and e turnover frequency at different overpotential values
Current-overpotential plots (Tafel plots) are used to further assess the kinetics and mechanism of OER [81]. The anodic polarization curves for OER were recorded in 1 M KOH and 1 M Methanol at the scan rate of 100 mV s−1. Tafel slopes were determined using the Tafel equation [82]:
where a is the Tafel constant measured in volts, b is the Tafel slope in mV dec−1, and J is the current density in mA.cm−2. The obtained Tafel slopes in 1 M KOH and 1 M KOH + 1 M Methanol are shown in Fig. 11b, c respectively, where the catalyst Ni30Fe70/CA shows the Tafel slope of 94.5 mV/dec (in 1 M KOH + 1 M Methanol), which is the lowest among all catalysts. Obtained Tafel slope value is also smaller than the previously reported catalysts. Such a small value of the Tafel slope indicates the more desirable OER kinetics for the as-prepared FeNi/CA catalysts. Calculated Tafel slopes for all electrocatalysts in both mediums and the change in Tafel slope upon the addition of methanol is shown in Table 6.
Standard current density (10 mA.cm−2) was chosen for the calculation of overpotential. A minimum overpotential of 608 mV was observed for the Ni30Fe70CA catalyst in 1 M KOH electrolyte. However, in the presence of methanol, overpotential was decreased remarkably (i.e. 377 mV at 10 mA.cm−2 current density) for this electrocatalyst. Overpotential in the presence and absence of methanol at 10 mAcm−2 current density is shown in Fig. 11d.
Turnover frequency (TOF) provides a precise description of the intrinsic properties of the catalysts. It is simply the measure of the ratio of the amount of product formed in a specific time to the amount of catalyst used. TOF for all the prepared electrocatalysts was calculated using the equation (TOF = i/4*F*m), where i is the current produced during the OER process, F is Faraday’s constant, and m is moles of catalyst. Calculated TOF for all electrocatalysts at different overpotential values is shown in Fig. 11e. The measured TOF for Ni70Fe30/CA is the highest among all prepared electrocatalysts (1.09 s−1 at 300 mV overpotential) and also high compared to previous reported catalysts, where Fe and Ni are used as efficiency booster [25, 83,84,85].
Yang and co-workers synthesized NiO-NiFe2O4 nanoparticles anchored on rGO support as an efficient OER electrocatalyst. As-synthesized nanohybrid electrocatalyst showed a low overpotential of 296 mV at a current density of 10 mA cm−2. This outstanding electrocatalytic activity was attributed to the interaction between the active NiO-NiFe2O4 and conductive rGO substrate [86]. Goodenough and co-workers prepared Ni3FeN with a large specific surface area, porous structure, high conductivity, and excellent efficiency towards OER. As-prepared catalyst delivered (η10 = 335 mV) overpotential and low Tafel slope of 70 mV dec−1 in alkaline media [87]. Some kinetic parameters of FeNi-based electrocatalysts for OER are summarized in Table 7.
4.2.4 Electrochemical impedance spectroscopy (EIS)
Electrochemical impedance spectroscopy was performed for the investigation of electron transfer capacities of prepared electrocatalysts for OER. 5 mv of AC voltage was supplied in addition to 1.1 to 1.5 V DC voltage in 1 M KOH and 1 M KOH + 1 M Methanol from 0.1 Hz to 20 k Hz of frequency. The resultant Nyquist plots were recorded and shown in Fig. 12.

a Nyquist plots for all electrocatalyst modified GC electrodes in 1 M KOH with 5 mV AC and 1.1 V DC voltage, b Nyquist plots for Fe/CA modified electrode in 1 M KOH with 5 mV AC, and (1.1–1.5 V) DC voltage, c Comparative Nyquist plots for all modified electrodes in 1 M KOH + 1 M Methanol with 5 mV AC and 1.1 V DC voltage, and d Comparison of Nyquist plots of Ni30Fe70/CA modified electrode in 1 M KOH and 1 M KOH + 1 M methanol with 5 mV AC and 1.1 V DC voltage. Inset represents the equivalent circuit model for parameter calculation
In Fig. 12a, comparative Nyquist plots in 1 M KOH at 1.1 DC voltage are presented. The Fe/CA modified electrode delivers the lowest charge transfer resistance, Rct (230.6). The EIS results and the electrocatalyst's CV response are in good agreement. In 1 M KOH (without methanol), Ni70Fe30/CA and Ni30Fe70/CA exhibited high resistance. But as can be shown in Fig. 9c, these materials possessed the lowest resistance when methanol was added. Figure 12b displays the Nyquist plot for the Fe/CA modified electrode at 5 mV AC voltage and 1.1 to 1.5 V DC voltage. Results showed that while charge transfer resistance (Rct) decreases as DC voltage is raised, solution resistance (Rs) is unaffected by the applied voltage. The electrode/electrolyte interface, which is a charge transfer barrier, is represented as an unfinished semicircle on the Nyquist plot of the OER reaction at low frequency. The semicircle at high frequency corresponds to the ionic resistance. The Nyquist plot clearly shows two types of resistance: ohmic resistance (Rs), caused by the resistance provided by the ionic electrolyte, and polarization resistance (Rct), which reflects the impediment provided by the charge transfer process. Warburg resistance (Rw), which is a straight line in the low-frequency domain, is considered to exist. To describe the not ideal behavior of capacitors, constant phase elements (CPE) were first developed. There are two parameters in it: and Q. In the absence of frequency dispersion, Q reflects the capacitive behavior, and is the exponent (0 ≥ α ≥ 1); for an ideal capacitor, α = 1 [98]. Figure 12d represents the comparative Nyquist plots in the presence and absence of methanol. With the addition of methanol, the diameter of the semicircle decreases, depicting ease in the electron transfer process. Charge transfer resistance decreased which is witnessed for high peak current, low onset and overpotential values [70]. Same results are obtained for the calculation of overpotential in the presence and absence of methanol shown in Fig. 11c. Using Eq. (8), the values of the apparent electron transfer rate constant for all the prepared electrocatalysts were estimated.
where R is the universal gas constant, T is the absolute temperature of the system, F is the Faraday constant and C is the concentration of the probe molecule.
EIS parameters are calculated from model fitting and represented in Table 8 recorded in 1 M KOH (a) and in 1 M KOH + 1 M methanol (b).
Nyquist plots show that upon increasing the DC voltage, the resistance of the electron transfer process decreases and upon the addition of methanol as well.
5 Conclusions
Sol–gel and microwave irradiation processes have been employed for the synthesis of Fe/CA and NixFe100-x/CA (x = 30, 50, 70). The physical and electrochemical properties of the prepared material were investigated. The high surface area and porous structure of the CA support material were supported by BET analysis, making it an ideal support for analyte species and transition metals. The variation of metal mass loading on CA support material was further supported by ICP-MS data. It can be seen that the synthesis was successful with the physical characterizations (XRD, XPS, SEM, EDAX, TEM, HR-TEM and SAED) made on the catalysts. An extensive electrochemical investigation shows that the produced electrocatalysts exhibit outstanding activity toward OER. Ni30Fe70/CA, the most effective electrocatalyst with a high diffusion coefficient (2.1 × 10−8 cm2s−1), mass transport coefficient (2.8 × 10−4 cm s−1), heterogeneous rate constant (6.1 × 10−4 cm s−1), and minimum Tafel slope (94.5 mV dec−1). The effect of methanol was remarkable on the electro-kinetics of OER. Ni30F70/CA showed very less overpotential when methanol was added. The high surface area obtained from CV also witnessed excellent activity of the catalyst. Stable electrocatalytic performance in alkaline media was observed over 3600 s. EIS analysis demonstrates low solution resistance (21.09 Ω), charge transfer resistance (10.18 Ω), and high apparent electron transfer rate constant (2.613 × 10−5 cm s−1) for Ni30Fe70/CA modified electrode. All materials demonstrated excellent OER activity and inspired the development of highly efficient NiFe-based catalysts for OER and opened the doors for numerous electrochemical applications.
Data availability
Data is available if required.
References
Yang A, Li T, Jiang S, Wang X, Qiu X, Lei W et al (2019) High-density growth of ultrafine PdIr nanowires on graphene: reducing the graphene wrinkles and serving as efficient bifunctional electrocatalysts for water splitting. Nanoscale 11:14561–14568
Du Z, Yang S, Li S, Lou J, Zhang S, Wang S et al (2020) Conversion of non-van der Waals solids to 2D transition-metal chalcogenides. Nature 577:492–496
Wu H, Chen Z, Wang Y, Cao E, **ao F, Chen S et al (2019) Regulating the allocation of N and P in codoped graphene via supramolecular control to remarkably boost hydrogen evolution. Energy Environ Sci 12:2697–2705
Zahedi R, Zahedi A, Ahmadi A (2022) Strategic study for renewable energy policy, optimizations and sustainability in Iran. Sustainability 14:2418
Lee JE, Jeon K-J, Show PL, Jung S-C, Choi YJ, Rhee GH et al (2022) Mini review on H2 production from electrochemical water splitting according to special nanostructured morphology of electrocatalysts. Fuel 308:122048
Chaudhary P, Ingole PP (2022) Electrocatalytic and photo-catalytic water splitting. In: Garg S, Chandra A (eds) Green photocatalytic semiconductors: recent advances and applications. Springer Nature, Berlin, pp 673–699
Li L, Wang P, Shao Q, Huang X (2020) Metallic nanostructures with low dimensionality for electrochemical water splitting. Chem Soc Rev 49:3072–3106
Zhang H, Maijenburg AW, Li X, Schweizer SL, Wehrspohn RB (2020) Bifunctional heterostructured transition metal phosphides for efficient electrochemical water splitting. Adv Func Mater 30:2003261
Liu Z, Zhao L, Liu Y, Gao Z, Yuan S, Li X et al (2019) Vertical nanosheet array of 1T phase MoS2 for efficient and stable hydrogen evolution. Appl Catal B 246:296–302
Ding Y, Cao K-W, He J-W, Li F-M, Huang H, Chen P et al (2022) Nitrogen-doped graphene aerogel-supported ruthenium nanocrystals for pH-universal hydrogen evolution reaction. Chin J Catal 43:1535–1543
Guan J, Bai X, Tang T (2022) Recent progress and prospect of carbon-free single-site catalysts for the hydrogen and oxygen evolution reactions. Nano Res 15:818–837
Zhang Q, Guan J (2021) Applications of atomically dispersed oxygen reduction catalysts in fuel cells and zinc–air batteries. Energy Environ Mater 4:307–335
Lopes PP, Chung DY, Rui X, Zheng H, He H, Farinazzo Bergamo Dias Martins P et al (2021) Dynamically stable active sites from surface evolution of perovskite materials during the oxygen evolution reaction. J Am Chem Soc 143:2741–2750
Li S, Gao Y, Li N, Ge L, Bu X, Feng P (2021) Transition metal-based bimetallic MOFs and MOF-derived catalysts for electrochemical oxygen evolution reaction. Energy Environ Sci 14:1897–1927
Feng C, Faheem MB, Fu J, **ao Y, Li C, Li Y (2020) Fe-based electrocatalysts for oxygen evolution reaction: progress and perspectives. ACS Catal 10:4019–4047
Kong X, Xu K, Zhang C, Dai J, Norooz Oliaee S, Li L et al (2016) Free-standing two-dimensional Ru nanosheets with high activity toward water splitting. ACS Catal 6:1487–1492
Huo J-M, Wang Y, Meng J, Zhao X-Y, Zhai Q-G, Jiang Y-C et al (2022) π⋯ π interaction directed 2D FeNi-LDH nanosheets from 2D Hofmann-MOFs for the oxygen evolution reaction. J Mater Chemi A 10:1815–1820
Han L, Dong S, Wang E (2016) Transition-metal (Co, Ni, and Fe)-based electrocatalysts for the water oxidation reaction. Adv Mater 28:9266–9291
Ma C, Sun W, Qamar Zaman W, Zhou Z, Zhang H, Shen Q et al (2020) Lanthanides regulated the amorphization–crystallization of IrO2 for outstanding OER performance. ACS Appl Mater Interfaces 12:34980–34989
Over H (2021) Fundamental studies of planar single-crystalline oxide model electrodes (RuO2, IrO2) for acidic water splitting. ACS Catal 11:8848–8871
Qin Y, Yang M, Deng C, Shen W, He R, Li M (2021) Theoretical insight into single Rh atoms anchored on N-doped γ-graphyne as an excellent bifunctional electrocatalyst for the OER and ORR: electronic regulation of graphitic nitrogen. Nanoscale 13:5800–5808
Cheng Y, Xu C, Jia L, Gale JD, Zhang L, Liu C et al (2015) Pristine carbon nanotubes as non-metal electrocatalysts for oxygen evolution reaction of water splitting. Appl Catal B 163:96–104
Zhao J, Liu Y, Quan X, Chen S, Zhao H, Yu H (2016) Nitrogen and sulfur co-doped graphene/carbon nanotube as metal-free electrocatalyst for oxygen evolution reaction: the enhanced performance by sulfur do**. Electrochim Acta 204:169–175
Zhao Y, Nakamura R, Kamiya K, Nakanishi S, Hashimoto K (2013) Nitrogen-doped carbon nanomaterials as non-metal electrocatalysts for water oxidation. Nat Commun 4:2390
Görlin M, Gliech M, de Araújo JF, Dresp S, Bergmann A, Strasser P (2016) Dynamical changes of a Ni-Fe oxide water splitting catalyst investigated at different pH. Catal Today 262:65–73
Ullal Y, Hegde AC (2014) Electrodeposition and electro-catalytic study of nanocrystalline Ni–Fe alloy. Int J Hydrogen Energy 39:10485–10492
Liu D, Yang Y, Zhang J, Wang L, Ma Z, Ren L et al (2023) Improved OER catalytic performance of NiFe-LDH with hydrothermal carbonization microspheres. J Alloy Compd 941:168994
Yu L, Zhou H, Sun J, Qin F, Yu F, Bao J et al (2017) Cu nanowires shelled with NiFe layered double hydroxide nanosheets as bifunctional electrocatalysts for overall water splitting. Energy Environ Sci 10:1820–1827
Ricciardi B, da Silva FW, Mecheri B, Nisa KU, Montero J, Ficca VC et al (2024) Hierarchical porous Fe/Ni-based bifunctional oxygen electrocatalysts for rechargeable zinc-air batteries. Carbon 219:118781
Huang S, Lu J, Wu X, Zhu H, Shen X, Cui S et al (2023) Ru-promoted NiFe oxyhydroxide anchored on the hierarchical porous N-doped carbon aerogel: Electronic structures modulation for much enhanced OER/HER dual-functional characteristics. Appl Catal A 664:119331
Zhang Y, Li Y, Yao Z, Wang J, Zhong Q (2022) Iron-nickel aerogels anchored on GO nanosheets as efficient oxygen evolution reaction catalysts under industrial conditions. Int J Hydrogen Energy 47:6996–7004
Lu J, Hao W, Wu X, Shen X, Cui S, Shi W (2023) Electronic modulation of the 3D architectured Ni/Fe oxyhydroxide anchored n-doped carbon aerogel with much improved OER activity. Gels 9:190
Chen Z, Zhang S, Yang J, Chen C, Song Y, Xu C et al (2021) High loading of NiFe active sites on a melamine formaldehyde carbon-based aerogel towards efficient bi-functional electrocatalysis for water splitting. Sustain Energy Fuels 5:4973–4980
Li H, Shu X, Tong P, Zhang J, An P, Lv Z et al (2021) Fe–Ni alloy nanoclusters anchored on carbon aerogels as high-efficiency oxygen electrocatalysts in rechargeable Zn–Air batteries. Small 17:2102002
Samancı M, Daş E, Bayrakçeken YA (2021) Carbon aerogel and their polypyrrole composites used as capacitive materials. Int J Energy Res 45:1729–1747
Daş E, Yurtcan AB (2016) Effect of carbon ratio in the polypyrrole/carbon composite catalyst support on PEM fuel cell performance. Int J Hydrogen Energy 41:13171–13179
Butt TM, Janjua NK, Mujtaba A, Zaman SA, Ansir R, Rafique A et al (2020) B-site do** in lanthanum cerate nanomaterials for water electrocatalysis. J Electrochem Soc 167:026503
Samancı M, Yurtcan AB (2022) Chemically and thermally reduced graphene oxide supported Pt catalysts prepared by supercritical deposition. Int J Hydrogen Energy 47:19669–19689
Hanzawa Y, Hatori H, Yoshizawa N, Yamada Y (2002) Structural changes in carbon aerogels with high temperature treatment. Carbon 40:575–581
Samancı M, Daş E, Yurtcan AB (2021) Effect of solvent exchange on the properties of carbon xerogel and carbon xerogel/polypyrrole composites for supercapacitors. Carbon Lett 31:1287–1308
Wu A, Yang X, Yang H (2012) Magnetic properties of carbon coated Fe, Co and Ni nanoparticles. J Alloy Compd 513:193–201
Hu Y (2021) A microwave radiation-enhanced Fe–C/persulfate system for the treatment of refractory organic matter from biologically treated landfill leachate. RSC Adv 11:29620–29631
Zhang Y, Liu C, Fan G, Yang L, Li F (2018) A robust core–shell nanostructured nickel–iron alloy@ nitrogen-containing carbon catalyst for the highly efficient hydrogenation of nitroarenes. Dalton Trans 47:13668–13679
Song L, Meng H (2010) Effect of carbon content on Ni–Fe–C electrodes for hydrogen evolution reaction in seawater. Int J Hydrogen Energy 35:10060–10066
Jiang N, Meng H-M, Song L-J, Yu H-Y (2010) Study on Ni–Fe–C cathode for hydrogen evolution from seawater electrolysis. Int J Hydrogen Energy 35:8056–8062
Ramsden A, Cameron E (1966) Kamacite and taenite superstructures and a metastable tetragonal phase in iron meteorites. Am Min J Earth Planet Mater 51:37–55
Zhang C, Li J, Shi C, Liu E, Du X, Feng W et al (2011) The efficient synthesis of carbon nano-onions using chemical vapor deposition on an unsupported Ni–Fe alloy catalyst. Carbon 49:1151–1158
Gopi S, Choi D, Ramu AG, Theerthagiri J, Choi MY, Yun K (2022) Hybridized bimetallic Ni–Fe and Ni–Co spinels infused N-doped porous carbon as bifunctional electrocatalysts for efficient overall water splitting. Int J Hydrogen Energy 52:190–201
Zhou Y, Zhang W, Hu J, Li D, Yin X, Gao Q (2021) Inherent oxygen vacancies boost surface reconstruction of ultrathin Ni-Fe layered-double-hydroxides toward efficient electrocatalytic oxygen evolution. ACS Sustain Chem Eng 9:7390–7399
Menzel M, Šepelák V, Becker K (2001) Mechanochemical reduction of nickel ferrite. Solid State Ionics 141:663–669
Gonzalez-Castano M, de Miguel JCN, Boelte J-H, Centeno MA, Klepel O, Arellano-Garcia H (2021) Assessing the impact of textural properties in Ni–Fe catalysts for CO2 methanation performance. Microporous Mesoporous Mater 327:111405
Karakaş ZK, Boncukcuoğlu R, Karakaş İH, Ertuğrul M (2015) The effects of heat treatment on the synthesis of nickel ferrite (NiFe2O4) nanoparticles using the microwave assisted combustion method. J Magn Magn Mater 374:298–306
Wan W, Zhao Y, Wei S, Triana CA, Li J, Arcifa A et al (2021) Mechanistic insight into the active centers of single/dual-atom Ni/Fe-based oxygen electrocatalysts. Nat Commun 12:5589
Feng Y, Yu X-Y, Paik U (2016) N-doped graphene layers encapsulated NiFe alloy nanoparticles derived from MOFs with superior electrochemical performance for oxygen evolution reaction. Sci Rep 6:34004
Yadav DK, Ganesan V, Sonkar PK, Gupta R (2017) Templated synthesis of nickel−iron layered double hydroxide for enhanced electrocatalytic water oxidation: towards the development of non-precious-metal catalysts. ChemElectroChem 4:3134–3139
Ni Y, Yao L, Wang Y, Liu B, Cao M, Hu C (2017) Construction of hierarchically porous graphitized carbon-supported NiFe layered double hydroxides with a core–shell structure as an enhanced electrocatalyst for the oxygen evolution reaction. Nanoscale 9:11596–11604
Liu YT, Tang L, Dai J, Yu J, Ding B (2020) Promoted electrocatalytic nitrogen fixation in Fe-Ni layered double hydroxide arrays coupled to carbon nanofibers: the role of phosphorus do**. Angew Chem Int Ed 59:13623–13627
Garibay Febles V, Melo-Máximo DV, Hernández Perez I, Suárez Parra R, Espinoza-Tapia JC, Luna Paz R et al (2022) Sonochemical synthesis of nanostructured Ni-Fe-C system and its catalytic activity based on decolorization of reactive black 5 dye. Crystals 12:1123
Fang D, He F, **e J, Xue L (2020) Calibration of binding energy positions with C1s for XPS results. J Wuhan Univer Technol Mater Sci Ed 35:711–718
Chen X, Wang X, Fang D (2020) A review on C1s XPS-spectra for some kinds of carbon materials. Fullerenes Nanotubes Carbon Nanostruct 28:1048–1058
Gao Z, Li A, Ma D, Zhou W (2022) Electron energy loss spectroscopy for single atom catalysis. Top Catal 65:1609–1619
Kang Q, Lai D, Tang W, Lu Q, Gao F (2021) Intrinsic activity modulation and structural design of NiFe alloy catalysts for an efficient oxygen evolution reaction. Chem Sci 12:3818–3835
Nasibulina LI, Koltsova TS, Joentakanen T, Nasibulin AG, Tolochko OV, Malm JE et al (2010) Direct synthesis of carbon nanofibers on the surface of copper powder. Carbon 48:4559–4562
Sayed FN, Polshettiwar V (2015) Facile and sustainable synthesis of shaped iron oxide nanoparticles: effect of iron precursor salts on the shapes of iron oxides. Sci Rep 5:9733
Wang Z, Liao X, Lin Z, Huang F, Jiang Y, Owusu KA et al (2020) 3D nitrogen‐doped graphene encapsulated metallic nickel–iron alloy nanoparticles for efficient bifunctional oxygen electrocatalysis. Chem A Eur J 26:4044–4051
Dominguez-Crespo MA, Ramírez-Meneses E, Montiel-Palma V, Huerta AT, Rosales HD (2009) Synthesis and electrochemical characterization of stabilized nickel nanoparticles. Int J Hydrogen Energy 34:1664–1676
Hu C, Zhang L, Gong JJE, Science E (2019) Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ Sci 12:2620–2645
Butt TM, Erum S, Mujtaba A, Medvedev D (2022) Janjua NKJFiC Nickel-doped lanthanum cerate nanomaterials as highly active electrocatalysts. Front Chem 10:1064906
Long J, Zhang J, Xu X, Wang F (2020) Crystalline NiFe layered double hydroxide with large pore volume as oxygen evolution electrocatalysts. Mater Chem Phys 254:123496
Roy A, Jadhav HS, Cho M, Seo JG (2019) Electrochemical deposition of self-supported bifunctional copper oxide electrocatalyst for methanol oxidation and oxygen evolution reaction. J Ind Eng Chem 76:515–523
Liu C, Feng L (2023) Advances in anode catalysts of methanol-assisted water-splitting reactions for hydrogen generation. Chin J Struct Chem 42:100136
Islam SE, Hang D-R, Liang C-T, Sharma KH, Huang H-C, Chou MM (2023) Trimetallic Ni–Co–Mo nanoparticles supported on N-doped carbon as a promising electrocatalyst for the methanol-assisted hydrogen evolution reaction. ACS Appl Energy Mater 6:9543–9555
Li M, Deng X, **ang K, Liang Y, Zhao B, Hao J et al (2020) Value-added formate production from selective methanol oxidation as anodic reaction to enhance electrochemical hydrogen cogeneration. Chemsuschem 13:914–921
Cui B, Wang C, Huang S, He L, Zhang S, Zhang Z et al (2020) Efficient multifunctional electrocatalyst based on 2D semiconductive bimetallic metal-organic framework toward non-Pt methanol oxidation and overall water splitting. J Colloid Interface Sci 578:10–23
Nisar L, Sadaqat M, Hassan A, Shah A, Najam-Ul-Haq M, Ashiq MN et al (2020) Ultrathin CoTe nanoflakes electrode demonstrating low overpotential for overall water splitting. Fuel 280:118666
Khan S, Shah SS, Anjum MAR, Khan MR, Janjua NK (2021) Electro-oxidation of ammonia over copper oxide impregnated γ-Al2O3 nanocatalysts. Coatings 11:313
Asim M, Hussain A, Khan S, Arshad J, Butt TM, Hana A et al (2022) Sol-gel synthesized high entropy metal oxides as high-performance catalysts for electrochemical water oxidation. Molecules 27:5951
Butt TM, Ullah A, Janjua NKJ (2022) Electrokinetic analysis of water oxidation on alumina supported silver oxide nanopowders. J Electroanal Chem 907:116053
Ashraf G, Asif M, Aziz A, Iftikhar T, Zhong Z-T, Zhang S et al (2022) Advancing interfacial properties of carbon cloth via anodic-induced self-assembly of MOFs film integrated with α-MnO2: a sustainable electrocatalyst sensing acetylcholine. J Hazard Mater 426:128133
Shi Y, Cai J, Zhang X, Li Z, Lin S (2022) Promotional effects of trace Ni on its dual-functional electrocatalysis of Co/N-doped carbon nanotube catalysts for ORR and OER. Int J Hydrogen Energy 47:7761–7769
Khalafallah D, Zhi M, Hong Z (2019) Recent trends in synthesis and investigation of nickel phosphide compound/hybrid-based electrocatalysts towards hydrogen generation from water electrocatalysis. Top Curr Chem 377:1–48
Bard AJ, Faulkner LR (2001) Fundamentals and applications. Electrochem Methods 2:482
Dionigi F, Reier T, Pawolek Z, Gliech M, Strasser P (2016) Design criteria, operating conditions, and nickel–iron hydroxide catalyst materials for selective seawater electrolysis. Chemsuschem 9:962–972
Gong M, Li Y, Wang H, Liang Y, Wu JZ, Zhou J et al (2013) An advanced Ni–Fe layered double hydroxide electrocatalyst for water oxidation. J Am Chem Soc 135:8452–8455
Long X, Li J, **ao S, Yan K, Wang Z, Chen H et al (2014) A strongly coupled graphene and FeNi double hydroxide hybrid as an excellent electrocatalyst for the oxygen evolution reaction. Angew Chem Int Ed 53:7584–7588
Zhang G, Li Y, Zhou Y, Yang F (2016) NiFe layered-double-hydroxide-derived NiO-NiFe2O4/reduced graphene oxide architectures for enhanced electrocatalysis of alkaline water splitting. ChemElectroChem 3:1927–1936
Fu G, Cui Z, Chen Y, Xu L, Tang Y, Goodenough JB (2017) Hierarchically mesoporous nickel-iron nitride as a cost-efficient and highly durable electrocatalyst for Zn-air battery. Nano Energy 39:77–85
Sun J-S, Zhou Y-T, Yao R-Q, Shi H, Wen Z, Lang X-Y et al (2019) Nanoporous gold supported chromium-doped NiFe oxyhydroxides as high-performance catalysts for the oxygen evolution reaction. J Mater Chem A 7:9690–9697
Liu H, Wang Y, Lu X, Hu Y, Zhu G, Chen R et al (2017) The effects of Al substitution and partial dissolution on ultrathin NiFeAl trinary layered double hydroxide nanosheets for oxygen evolution reaction in alkaline solution. Nano Energy 35:350–357
Yang Y, Dang L, Shearer MJ, Sheng H, Li W, Chen J et al (2018) Highly active trimetallic NiFeCr layered double hydroxide electrocatalysts for oxygen evolution reaction. Adv Energy Mater 8:1703189
Weng B, Xu F, Wang C, Meng W, Grice CR, Yan Y (2017) A layered Na1− xNiyFe1− yO2 double oxide oxygen evolution reaction electrocatalyst for highly efficient water-splitting. Energy Environ Sci 10:121–128
Jia X, Zhao Y, Chen G, Shang L, Shi R, Kang X et al (2016) Ni3FeN nanoparticles derived from ultrathin NiFe-layered double hydroxide nanosheets: an efficient overall water splitting electrocatalyst. Adv Energy Mater 6:1502585
Wu C, Zhang X, **a Z, Shu M, Li H, Xu X et al (2019) Insight into the role of Ni–Fe dual sites in the oxygen evolution reaction based on atomically metal-doped polymeric carbon nitride. J Mater Chem A 7:14001–14010
Hu Q, Liu X, Zhu B, Fan L, Chai X, Zhang Q et al (2018) Crafting MoC2-doped bimetallic alloy nanoparticles encapsulated within N-doped graphene as roust bifunctional electrocatalysts for overall water splitting. Nano Energy 50:212–219
Wang CP, Liu HY, Bian G, Gao X, Zhao S, Kang Y et al (2019) Metal-layer assisted growth of ultralong quasi-2D MOF nanoarrays on arbitrary substrates for accelerated oxygen evolution. Small 15:1906086
Shi Y, Yu Y, Liang Y, Du Y, Zhang B (2019) In situ electrochemical conversion of an ultrathin tannin nickel iron complex film as an efficient oxygen evolution reaction electrocatalyst. Angew Chem Int Ed 58:3769–3773
Kumar A, Bhattacharyya S (2017) Porous NiFe-oxide nanocubes as bifunctional electrocatalysts for efficient water-splitting. ACS Appl Mater Interfaces 9:41906–41915
Alobaid A, Wang C, Adomaitis RA (2018) Mechanism and kinetics of HER and OER on NiFe LDH films in an alkaline electrolyte. J Electrochem Soc 165:J3395–J3404
Acknowledgements
Muhammad Asim and Akbar Hussain are gratefully acknowledged for Türkiye Scholarships.
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Asim, M., Hussain, A., Samancı, M. et al. Carbon aerogel supported Ni–Fe catalysts for superior oxygen evolution reaction activity. Carbon Lett. (2024). https://doi.org/10.1007/s42823-024-00730-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42823-024-00730-4