
Abstract
SARS-CoV-2 emerged in 2019 and led to the COVID-19 pandemic. Efforts to develop therapeutics against SARS-Cov-2 led to both new treatments and attempts to repurpose existing medications. Here, we provide a narrative review of the xenobiotics and alternative remedies used or proposed to treat COVID-19. Most repositioned xenobiotics have had neither the feared toxicity nor the anticipated efficacy. Repurposed viral replication inhibitors are not efficacious and frequently associated with nausea, vomiting, and diarrhea. Antiviral medications designed specifically against SARS-CoV-2 may prevent progression to severe disease in at-risk individuals and appear to have a wide therapeutic index. Colloidal silver, zinc, and ivermectin have no demonstrated efficacy. Ivermectin has a wide therapeutic index but is not efficacious and acquiring it from veterinary sources poses additional danger. Chloroquine has a narrow therapeutic index and no efficacy. A companion review covers vaccines, monoclonal antibodies, and immunotherapies. Together, these two reviews form an update to our 2020 review.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a coronavirus that causes coronavirus disease 2019 (COVID-19), a respiratory infection characterized by flu-like symptoms and can lead to myocarditis, acute respiratory distress syndrome (ARDS), and death [1].
While waiting for vetted therapeutics, an increasingly desperate population turned to remedies with little to no supporting evidence, including hydroxychloroquine, ivermectin, and zinc. Since 2019, many FDA-approved medications and substances not approved by the FDA have been posited or repositioned to treat COVID-19. Since 2020, vaccines, monoclonal antibodies, and antivirals have been developed for prophylaxis and the prevention of progression to severe disease.
This narrative review describes the potential toxicities of xenobiotics that a clinician is likely to encounter when treating patients affected by COVID-19. Clinicians may encounter the use of repositioned medications that were ultimately found to be ineffective. A companion manuscript discusses biological therapeutics, such as convalescent plasma, monoclonal antibodies, and vaccines. We intend for these reviews to familiarize readers with the toxic effects of COVID-19 therapeutics and how to address the toxicities, only touching on efficacy. The phrase “we previously discussed” refers to our 2020 review [2] unless otherwise stated.
Methodology
For each substance we previously discussed, we searched PubMed and Google Scholar for any publications between May 1, 2020, and April 1, 2022, that mentioned the substance and contained the keywords “adverse event,” “secondary effect,” “unintended effect,” “adverse reaction,” “unintended reaction,” or “toxicity,” or their spelling variants, in the full text of the paper. We considered papers tagged with MeSH Heading “Drug Toxicity” (D064420), also labeled as “Adverse Drug Event,” and all subheadings. This MeSH heading also includes all adverse drug reactions, which are the intersection of the MeSH tags D064420 and D004347 (“Drug Interaction”). We only considered peer-reviewed publications for which the full text in English was available.
For substances we did not previously discuss, we followed our earlier approach of choosing the substances to include based on discussion in the media, trends in calls to Poison Control Centers, and our experience as practicing physicians and scientists. We investigated two types of information for each substance. We relied on news articles, social media posts, commentaries, preprints, and peer-reviewed research to describe the substance’s rise to prominence in national discussions and perceived effects. We searched PubMed and Google Scholar for publications mentioning the substance published before June 1, 2022, to identify the substance’s formally reported effects. We only considered manuscripts with the full text available in English. We indicate the source with in-text and numerical citations to distinguish anecdotes from peer-reviewed publications. We consider preprints that were not linked to a peer-review article that is published or in press as anecdotal. To identify news articles and social media posts, we used the same Google Scholar search terms but restricted our results to news articles and social media posts.
Our description of substances and their effects is not exhaustive and presents our knowledge as of June 2022. In the absence of direct evidence, we expect drugs of a similar class to have similar toxicity. Our search strategy may be biased towards mild and moderate effects, which are assiduously reported by clinical trials. Case reports often provide the first description of outstanding cases of toxicity, but the cause of toxicity may not be identified.
Relevant Biology
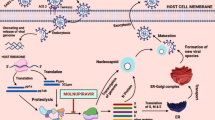
A deeper understanding of how SARS-CoV-2 enters cells and replicates has led to the development of inhibitors of viral entry and replication specific to SARS-CoV-2.
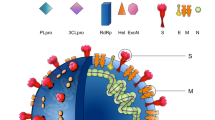
SARS-CoV-2 enters human respiratory epithelial cells when its Spike (S) protein interacts with the angiotensin-converting enzyme 2 (ACE2) receptor and adjacent transmembrane protease serine 2 (TMPRSS) on the host cell membrane. A host cell protease cleaves the S protein into two fragments, S1 and S2. The C-terminal region of S1 binds to ACE2 and is the target of neutralizing antibodies. The N-terminal region of S1 governs how strongly the S protein binds to cells. TMPRSS cleaves S2, allowing the plasma membranes of SARS-CoV-2 and the host cell to fuse. The replication of SARS-CoV-2 can trigger an inflammatory response that leads to acute lung injury and respiratory failure, the main cause of death from COVID-19 [3].
Chloroquine and Hydroxychloroquine
In early 2020, chloroquine and hydroxychloroquine were posited to reduce SARS-CoV-2 entry and dampen cytokine release. Our previous article discussed the risk of hypokalemia and cardiac dysrhythmias from chloroquine and hydroxychloroquine at daily doses of 4 to 5 g. One peer-reviewed clinical trial [4] and one preprint of a clinical trial [5] suggested daily doses of 1 g chloroquine and 600 mg hydroxychloroquine. Hydroxychloroquine is dosed at 200 to 400 mg each day when used to treat connective tissue disorders. No new or unanticipated toxicities have been reported.
A meta-analysis in 2021 of 14 published and 14 unpublished trials (total 10,319 inpatients) found that those who received chloroquine were 10% more likely to die at 28 days than those who received neither chloroquine nor hydroxychloroquine; those who received hydroxychloroquine had no increased risk [6]. Adverse events reported to the FDA associated with hydroxychloroquine more than doubled in 2022 as compared with 2018 or 2019 [7]. The distribution of types of adverse reactions (e.g., cutaneous, cardiac) was similar across the years, but the fraction of fatal adverse events was higher in 2020 (5.1%) than in 2019 or 2018 (1.9% and 3.1%). We direct readers to our prior review for further discussion, including treatment.
Viral Replication Inhibitors
SARS-CoV-2 must co-opt the host machinery to translate its RNA genome into viral proteins. It translates its genome using virally encoded RNA-dependent RNA polymerase (RdRp). RdRp is a highly conserved enzyme in positive-sense RNA viruses [8]. In the USA, the nucleotide analog molnupiravir (Lagevrio, manufactured by Merck) and protease inhibitor nirmatrelvir/ritonavir (Paxlovid, manufactured by Pfizer) are authorized by the Food and Drug Administration (FDA) under an emergency use authorization (EUA). The nucleotide analog remdesivir is approved by the FDA to treat COVID-19. All three prevent RdRp from faithfully translating the viral genome of SARS-CoV-2.
Nucleotide Analogs
Nucleotide analogs aim to prevent infected host cells from replicating the viral genome by halting the production of new viral RNA (vRNA). These analogs trick RNA polymerases to incorporate nucleotide variants that physically obstruct polymerase attachment or vRNA chain elongation into the replicating strand. For example, remdesivir incorporates itself into vRNA physically distorting the genome and creating a roadblock for RdRp [9].
Toxicity from nucleoside analogues arises from interference with mitochondrial DNA-dependent RNA polymerase [10]. Signs of toxicity include hyperlactatemia, peripheral neuropathy, bone marrow suppression, myopathy, and pancreatitis. In our prior paper, we suspected remdesevir would be well tolerated except in large overdose, with metabolic acidosis being the primary toxicity. In practice, remdesivir has been associated with bradycardia, transaminase elevation, and reduced renal function. Favipiravir has been associated with hyperuricemia and transaminase elevation. Molnupiravir has few adverse effects but is not recommended in pregnant patients due to concern for fetal harm.
Remdesivir
Remdesivir (Velkury, manufactured by Gilead) is a prodrug metabolized to an analog of adenosine. A meta-analysis of 5 clinical trials (of 395 screened) conducted up to July 2021 reported that those who received 10 days of remdesivir reported nausea, vomiting, and diarrhea more frequently than those who received other treatments (10% vs 4%) [11]. A phase 3 trial reported that nausea (37 of 384 vs 6 of 200), hypokalemia (23 of 384 vs 4 of 200), and headache (20 of 384 vs 5 of 200) were more common in those who received remdesivir than other treatments; a reduction to a creatinine clearance of less than 30 mL/minute was less frequent in those who received 5 days of remdesivir than those who received other treatments (71 of 354 vs 55 of 183), but more common in those who received 10 days of remdesivir (45 of 176) versus 5 days (26 of 178) [12]. In our prior review, we speculated that hyperlactatemia would be observed, but this has not been reported.
Compared to 115 patients with no renal impairment, 20 patients with pre-existing renal impairment were more likely to have transaminase elevations and worsening renal function when given remdesivir, although the authors reported no significant association between remdesivir and worsening organ function [13]. Post-marketing surveillance from February 1 to August 7, 2020, reported 138 cases of acute renal failure (out of 363 cases of acute renal injury) after remdesivir administration as opposed to 138 cases for tocilizumab, hydroxychloroquine, and lopinavir/ritonavir combined [14]. Decreased renal function could lead to the accumulation of sulcobutylether-beta-cyclodextrin (SBECD), an excipient used with remdesivir. Accumulation of SBECD is associated with renal tubular vacuolation and foamy macrophages in the liver and lungs in animals [15]. Patients who received 6 g SBEDC per 100 mg remdesivir did not have to stop treatment due to adverse events more than those who received 3 g per 100 mg (0.9% versus 2.3%) [16].
Bradycardia has been reported after remdesivir [17,18,19]. For example, two people who received remdesivir, and other medications including azithromycin and anakinra, developed bradycardia (one on the fourth day of treatment and one a “few” days into treatment) that resolved within 24 h of stop** remdesivir [20]. How remdesivir causes bradycardia is unknown. As an adenosine analog, remdesivir or its metabolites could reduce sinus node automaticity through vagal stimulation. Most cases resolve without intervention. To treat symptomatic bradycardia, atropine or a vasopressor with β-agonism would be appropriate. One 72-year-old male developed atrial fibrillation with a rapid ventricular response on the first day of a remdesivir infusion and a self-resolving complete heart block on the fifth day of infusion [21].
One patient developed blisters after extravasation of remdesivir [22]. Three patients in whom remdesivir infusions extravasated on the back of their palms experienced swelling, pain, and, in one case, a hematoma, all of which resolved within 1 week without intervention. Other infiltrations resulted in swelling or pain [23]. Local infiltration can be managed, as with most other extravasations, with warm compresses and, if needed, hyaluronidase.
Favipiravir
Favipiravir (Avigan, manufactured by Toyama Chemical) is an adenosine and guanine (purine) analog that leads RdRp to make nonfunctional viral proteins [24]. Favipiravir is used in Japan, Russia, Serbia, India, Turkey, Thailand, Egypt, and Hungary to treat COVID-19. It is not approved in the USA or the UK. A meta-analysis of 15 clinical trials found hyperuricemia and increased alanine aminotransferase more common in patients receiving favipiravir than controls (relative risk 7.69 and 1.35, respectively) [25].
An asymptomatic doubling of serum uric acid levels that resolved within 28 days occurred in 41.7% of patients (45/108) receiving favipiravir in a phase III clinical trial [26]. In patients suffering from monosodium urate crystalline arthropathy (gout), it may be appropriate to prefer other viral replication inhibitors. Favipiravir leads to increased first-trimester fetal demise and delayed development in mice, rats, rabbits, and monkeys [27].
Molnupiravir
Molnupiravir (MK-4482), manufactured by Merck and Ridgeback, is an oral prodrug of a ribonucleoside analog, N4-hydroxycytidine. Molnupiravir can substitute for uridine or cytidine, introducing C-to-U transitions and G-to-A transcription errors [28]. As transcription errors accumulate, each replication cycle yields fewer functional proteins, ultimately leading to the production of completely nonfunctional virions [29]. Introducing some transcription errors, but not enough to generate completely nonfunctional proteins, could prod SARS-CoV-2 to develop resistance to C-to-U and G-to-A substitutions. This has not been observed in vitro or in vivo.
N4-hydroxycytidine did not impair oxidative phosphorylation in HepG2 cell assays [30], a cell line commonly used in drug metabolism and hepatotoxicity studies. HepG2 cells require mitochondrial function (i.e., oxidative phosphorylation) if the incubation media do not contain glucose [31].
In dogs, 7 days of 17 mg/kg/day or 50 mg/kg/day of molnupiravir resulted in mild pancytopenia that spontaneously resolved after one month for the dogs who received 17 mg/kg/day. The dogs who received 50 mg/kg/day were euthanized 10 days after treatment ended. More severe thrombocytopenia and associated hemorrhage was observed in dogs receiving molunpiravir continuously for 14 to 21 days [32].
The MOVe-OUT trial was a phase 3 double-blind, randomized, placebo-controlled trial to evaluate the efficacy and safety of molnupiravir started within 5 days after the onset of signs or symptoms in nonhospitalized, unvaccinated adults with mild-to-moderate, laboratory-confirmed COVID-19 and at least one risk factor for severe COVID-19 illness. In that trial, 216 of 710 patients who received molnupiravir and 231 of 701 who received a placebo (30.4% and 33.0%, respectively) experienced one or more adverse events, most frequently diarrhea, nausea, and dizziness [33]. One out of 710 developed thrombocytopenia (platelet count < 50,000), which occurred on day 12 of the study. 6.3% of patients treated with molnupiravir vs 9.6% in the placebo group developed pneumonia [34]. No acute overdoses were reported. Serum lactate concentrations were not reported.
At the time of writing, the Food and Drug Administration (FDA) recommends that pregnant women do not use molnupiravir [35]. Pregnant rats and rabbits exposed to molnupiravir during organogenesis had pups with low birth weight, decreased bone density, and more frequent fetal demise [36]. Pregnant women and women who declined contraception were excluded from the MOVe-OUT trial. No human data are available for review.
Protease Inhibitors
Protease inhibitors, a class of drugs historically used to treat hepatitis C and HIV, prevent infected cells from generating new infectious virions by binding to and inactivating viral proteases. Viral proteases process the initial products of translation into functional viral proteins. Each virus has its own unique proteases. Drugs that target one viral protease may not inhibit another virus’ protease. For example, protease inhibitors that target HIV protease do not target the Hepatitis C NS3 protease [37, 38].
Lopinavir-Ritonavir
Ritonavir inhibits cytochrome CYP 3A4, the enzyme that inactivates lopinavir [39]. It is a potent inhibitor of CYP3A4 (Ki 0.59 ± 0.12 micromolar) and may slow the metabolism of drugs that are substrates of CYP3A4 [40]. Polymorphisms in CYP3A4 can make patients even more susceptible to drug-drug interactions.
Gastrointestinal symptoms are common and can prevent patients from completing treatment with lopinavir-ritonavir. In a randomized controlled open-label trial of 400 mg lopinavir-100 mg ritonavir twice a day for 14 days in patients hospitalized with confirmed SARS-CoV-2 infection and SpO2 less than or equal to 94%, there was no difference in time to clinical improvement or 28-day all-cause mortality between those who received lopinavir-ritonavir or placebo. However, 13 of 99 patients who received lopinavir-ritonavir had to stop treatment early because of severe nausea, vomiting, and diarrhea [41]. Three out of 10 patients in Wuhan, China, who received lopinavir, interferon alpha, and unspecified antibiotics experience nausea, vomiting, diarrhea, and serum potassium levels lower than 3.5 mmol/L that resolved after stop** lopinavir [42]. In the first published case series of COVID-19 infections in Singapore (in 2020), 5 of 18 patients received 400 mg lopinavir-100 mg ritonavir twice a day for 14 days but only 1 of 5 completed all 14 days; 4 of 5 developed nausea, vomiting, and diarrhea and 3 of 5 developed abnormal liver function tests not described further in the manuscript [43].
In those being treated for HIV with lopinavir-ritonavir, which uses the same 400 mg to 100 mg dosage, 3–10% develop increases in transaminases after a month of treatment that resolve with stop** the medication and are not associated with clinically significant liver dysfunction [44]. Lopinavir-ritonavir is typically administered for 14 days to treat COVID-19. Arthralgia [45] and Achilles tendinopathy [46] have been reported in individuals taking lopinavir-ritonavir, even in short-term use for HIV post-exposure prophylaxis.
Nirmatrelvir
Nirmatrelvir (PF-07321332) is an oral protease inhibitor marketed in combination with ritonavir as Paxlovid™ by Pfizer. It targets the 3-chymotrypsin-like-protease (3CLpro, also called the main protease, Mpro) of SARS-Cov-2. Mpro is an attractive pharmacologic target because it does not resemble human proteases and is conserved across viral variants. As with lopinavir, ritonavir decreases the metabolism of nirmatrelvir by inhibiting CYP3A4. Computer simulations suggest that nirmatrelvir binds more avidly to 3CLpro than does lopinavir-ritonavir [47]. In vitro, nirmatrelvir is a potent inhibitor of 3CLpro with EC50 and EC90 values (the concentrations required to produce 50 and 90% of maximal enzyme response, a marker of potency) of 77.9 and 215 nM in A549-ACE2 cells [48]. Nirmatrelvir maintained antiviral potency against new variants of concern in vitro, including omicron [38].
In phase 2/3 clinical trials dysgeusia occurred in 62 of 1109 (5.6%), compared with 3 of 1115 (0.3%) in the control group, diarrhea in 34 (3.1%) vs 18 (1.6%), and vomiting in 12 (1.1%) vs 9 (0.8%). Serious adverse events occurred in 1.6% of patients receiving nirmatrelvir, compared with 6.6% of the placebo group; COVID-19 pneumonia (6 of 1109 vs 37 of 1115), worsening COVID-19 symptoms (2 of 1109 vs 8 of 1115), and decreased creatinine clearance (2 of 1109 vs 3 of 1115) were the most common serious adverse events. No other clinically significant laboratory abnormalities were reported. Clinically significant liver toxicity was not observed. No patients receiving nirmatrelvir died in this trial; 12 patients who received the placebo did [49].
Ritonavir
Ritonavir inhibits CYP 3A4, CYP2D6, CYP2C19, and CYP2C9. In the context of treating COVID-19, it is used only as an adjuvant to decrease the metabolism of other protease inhibitors. The main clinical concern with ritonavir use is a drug-drug interaction. CYP3A4 is the most abundant cytochrome P450 in the liver, responsible for the metabolism of 50–60% of all pharmaceuticals including antiarrhythmics, antihypertensives, opioid analgesics, ergotamines, immunosuppressants, and antidepressants [50]. Patients most likely to benefit from remdesivir are also more likely to have comorbidities requiring medications that are metabolized by 3A4 or 2D6. The risk of stop** a patient’s usual medication must be weighed against the threat of severe COVID-19 illness. The dose of tacrolimus or cyclosporine may need to be paused or reduced, guided by obtaining serum drug concentrations [51]. Adjusting dosage may be more difficult for drugs whose concentrations are not readily measured or whose serum concentrations do not reflect tissue concentration. Consultation with a pharmacist or medical toxicologist may help minimize drug-drug interactions. The National Institutes of Health (NIH) maintains a list of medications, categorizing by severity of interactions, along with advice on whether to continue, hold, or reduce a patient’s usual medication. Online tools and calculators are also available to determine interactions, such as https://www.covid19-druginteractions.org/checker.
Of 350 adults with HIV who received ritonavir for 60 weeks, 35 (10.0%) experienced an increase in total cholesterol above 200 mg/dL compared with 10 out of 303 (3.3%) who did not [52]. At doses up to 100 mg/kg/day, there were no effects on male and female fertility or embryonic development in the first trimester in rats or rabbits [53]. Since treatment for COVID-19 is typically 5 to 14 days for most medications containing ritonavir, we do not anticipate significant changes in lipid profiles or fertility. The structural difference between human proteases and 3CLpro and the specificity of nirmatrelvir for 3CLpro suggest that off-target effects are unlikely. At the time of writing, there are no human data available for review.
It is reasonable to treat toxicity arising from drug-drug interactions by stop** the use of xenobiotics containing ritonavir. It is not advisable to start 3A4 inducers, such as phenobarbital, phenytoin, rifampicin, St. John’s Wort, or glucocorticoids because of the side effects associated with these medications.
Less Targeted Xenobiotics
Dexamethasone
Dexamethasone is a glucocorticoid with broad anti-inflammatory activity often used to decrease inflammation in reactive airway disease. It was proposed that dexamethasone would attenuate the body’s proinflammatory response, which SARS-CoV-2 seemed to elicit more strongly than other viruses. A multicenter open-label trial in England (RECOVERY) comparing 2104 patients with 4321 controls found that 6 mg of dexamethasone once daily for 10 days reduced the death rate at 28 days by 17% (2.7% absolute reduction) in patients hospitalized with COVID-19 who were develo** acute respiratory distress syndrome, with patients on mechanical ventilation benefitting the most [54, 55].
The main acute toxicities from dexamethasone are mania, dyslipidemia, and decreased glucose sensitivity. Psychiatric adverse effects occur in 1.3% of patients receiving 40 mg of prednisone each day (equivalent in anti-inflammatory effect to 6 mg dexamethasone) and 18.4% of patients receiving 80 mg prednisone (12 mg dexamethasone) each day[56, 57]. Usually, no treatment is needed other than to keep the patient safe (in the case of mania) and control glucose until the medication clears. Dexamethasone induces CYP3A4, opposite to ritonavir.
Azithromycin
Azithromycin is a macrolide antibiotic. At doses greater than 500 mg daily, azithromycin inhibits the hERG-mediated repolarizing current, IKr, which can lead to QTc prolongation and cardiac dysrhythmias [58]. We previously hypothesized that the combination of azithromycin and chloroquine or hydroxychloroquine would be more likely to precipitate cardiac dysrhythmias than either alone because chloroquine and hydroxychloroquine also inhibit hERG [59]. We could find no data confirming or denying this hypothesis, except from one group in France, referenced below, whose methods and findings have been formally questioned. In a cohort study of 90 patients hospitalized for COVID-19 who received azithromycin or hydroxychloroquine, those who received both had a median (interquartile range) change in QT interval of 23 (10 to 40 ms) compared with those who received only hydroxychloroquine 5.5 (− 15.5 to 34.2 ms) [60].
An open-label prospective trial in France of 22 patients reported that 200 mg hydroxychloroquine 3 times a day for 10 days and azithromycin 500 mg on the 1st day and then 250 mg each day for the next 4 days reduced viral load more than hydroxychloroquine alone [4]. This trial broke randomization. Those who declined treatment were analyzed as controls. A retrospective analysis of 3737 patients by the same group, which may have included patients from the previously mentioned prospective trial, found that 0.67% (25 patients) who received the dosing regimens described above had an increase of more than 60 ms in their QTc [61]. This study did not comment on the number of dysrhythmias or the association between the QTc increase and dysrhythmias.
ACE Inhibitors
In 2019 and early 2020, there was a concern raised on social media that ACE inhibitors could increase susceptibility to SARS-CoV-2 [62]. ACE inhibitors can increase ACE2 expression in human type II alveolar cells [63], which could create more binding sites for SARS-CoV-2. A retrospective single-center study from Wuhan, China, in 2020 identified hypertension, diabetes, and cerebrovascular disease as poor prognostic factors, but did not isolate the risk attributable to taking an ACE inhibitor [64]. These reports created concern that patients would discontinue a beneficial medication, although no study to date has reported on changes in adherence because of COVID.
Subsequent studies have found no association between ACE inhibitors or angiotensin receptor blockers (ARBs) and mortality from COVID-19 [65]. A pooled analysis (10 studies, 49,188 patients) found that SARS-CoV-2-positive patients taking ACEIs or ARBs were not more likely to die (RR 0.89; 95% CI 0.64–1.23; P = 0.48) or have worse disease (RR 1.29; 95% CI 0.81–2.04; P = 0.28) than those not taking ACEIs/ARBs [66].
Ivermectin
Ivermectin (Stromectol), manufactured by Merck, is a highly active broad-spectrum antiparasite medication that increases chloride influx by binding to glutamate-gated chloride channels, paralyzing organisms by preventing depolarization [67]. Arthropod glutamate-gated chloride channels resemble human GABA-A and glycine channels. Ivermectin, in vivo, is a substrate for human p-glycoprotein efflux pumps, which may explain why ivermectin is more toxic to worms than humans [68].
In 2020, an Australian research group reported that a single dose of ivermectin inhibited RNA replication in vitro of the SARS-CoV-2 variant then dominant in Australia (betaCoV/Australia/VIC01/2020)[69]. Social media sites then promoted ivermectin as efficacious against COVID-19 in humans, leading to a run on veterinary stock in the UK and illegally importing the drug from unlicensed foreign distributors [70]. One month later, the FDA warned against using ivermectin noting that the study did not determine whether ivermectin treated COVID-19 in humans[71]. Antiviral Research, the journal that published the in vitro study, published a letter to the editor 2 weeks later noting that humans would have to consume 10 times the dose approved by the FDA to achieve the intracellular concentration in the in vitro study [72]. A meta-analysis of 15 clinical trials initially found that ivermectin prophylaxis reduced COVID-19 infections by 79–91%, but this study was later qualified by an editorial Expression of Concern because 2 of the 15 trials included in the analysis were retracted [73]. We refer the reader to [74] for a detailed timeline of social media discussions and official actions related to ivermectin in the COVID-19 pandemic.
Veterinary ivermectin contains microcrystalline cellulose, pregelatinized maize starch, magnesium stearate, butylated hydroxyanisole, and citric acid as excipients. There is no evidence for toxicity of magnesium stearate [75] or butylated hydroxanisole [76] in the level allowed as food additives. That level is about 10% of the level in veterinary ivermectin [77]. We expect pregelatinized maize starch and microcrystalline cellulose to act as a bulk laxative.
The dose of ivermectin to treat parasitic infections is 0.2 mg/kg three times a week. A study of 68 healthy human volunteers recorded no adverse events in dosing up to 2 mg/kg [78]. 10 mg/kg of ivermectin, but not 5 mg/kg, induced generalized seizures in 12 wild-type mice that could be stopped with 5 mg/kg diazepam [79]. Of 164 patients hospitalized with confirmed mild COVID-19, those who received 12 mg ivermectin each day for 3 days stayed 8.82 ± 4.94 days in the hospital, compared to 10.97 ± 5.28 days for those who did not, a statistically insignificant difference [80]. Another study that used one dose of 0.2 mg/kg reported 173 hospitalized patients treated with ivermectin had lower mortality than 107 patients who did not receive ivermectin; unmatched control OR was 0.29–0.96, p = 0.03 (matched cohort 0.045, although there was no difference in length of stay or time spent on mechanical ventilation [81]).
From January through July 2021, the Oregon Poison Control Center reported that the rate of calls regarding ivermectin increased from 0.25 calls per month in 2020 to 0.86 calls per month, spiking in August 2021 to 21 calls of which 6 required hospitalization and 4 admission to the ICU for hypotension or ataxia [82].
It is unclear how ivermectin ingestion causes hypotension. It could inhibit sympathetic outflow, as benzodiazepines or barbiturates do, or have a more specific effect, like inhibiting sarcolemmal L-type calcium channels as midazolam or diazepam do [83, 84]. In human embryonic kidney cells, ivermectin binds to GABA-A channels with an EC50 of approximately 1 μM. Lorazepam’s in vitro EC50 at the same channel is 0.28 μm [85].
There is no antidote to ivermectin. If the patient can tolerate medication by mouth and ingested ivermectin within the last 2 h, we recommend 1 g/kg of activated charcoal. The time frame of 2 h is based on expert opinion. Flumazenil is unlikely to reverse hypotension from ivermectin. Flumazenil binds to the GABA-A receptor at the benzodiazepine binding site, the interface between α and γ subunits [86]. Crystal structures and in vitro site-directed mutagenesis studies suggest that ivermectin binds between the β and γ subunits [87].
Nutritional Supplements
Zinc
Zinc (Zn) plays a critical role in supporting the normal development and function of innate and adaptive immunity. In 2020, social media proposed zinc to treat COVID-19 [88]. A pathologist’s blog post in March 2020 [89] was distorted into a meme suggesting zinc as a cure-all [90]. Social media also referenced a pooled analysis in 2012 sponsored by Bayer Pharmaceuticals that reported that 5 days of 1000 mg of ascorbic acid and 10 mg of zinc each day decreased the severity of a runny nose in those with the “common cold” [91]. That study did not specify the form of zinc administered, nor did it confirm infection with a coronavirus. A randomized clinical trial of 214 outpatients with confirmed SARS-CoV-2 reported that symptoms lasted 5–7 days no matter whether patients received 10 days of daily 50 mg zinc gluconate (12.5 mg elemental zinc), 8000 mg ascorbic acid, both agents, or neither [92].
Acute toxicity from zinc ingestion, as with most metals, arises from its corrosive effects on the gastrointestinal tract, leading to nausea, vomiting, and epigastric pain. Long-term use above the maximum tolerable limit is associated with recurrent infections, decreased high-density lipoprotein cholesterol levels, sideroblastic anemia, granulocytopenia, an increased incidence of kidney stones, and copper deficiency [93]. One patient developed neutropenia and an undetectable copper level after consuming 50 mg of elemental zinc each day, requiring hospitalization and filgrastim [94]. The maximum tolerable limit of elemental zinc in healthy adults who are not pregnant is 40 mg per day.
The diagnostic assessment of total body zinc is challenging. Whole-blood concentration more accurately reflects total body zinc than serum concentration does. Zinc accumulates in red blood cells. Spot urine levels have no utility in estimating the total body burden of zinc. Twenty-four-hour urine tests must be collected in an acid-washed urinal and then extracted with polythiocarbamate to prevent zinc from binding to the container, requiring specialized equipment and expertise [50]. The most judicious approach to diagnosis and treatment is to recognize signs and symptoms of excess (anemia, gastrointestinal distress, sensory ataxia and spasticity, recurrent kidney stones) and stop the exposure.
Those with chronic zinc exposure may demonstrate spasticity or sensory ataxia on physical exam and T2 hyperintensity in the dorsal column, like B12 deficiency. This T2 hyperintensity differentiates zinc toxicity from copper deficiency, which is only associated with bilateral subcortical T2 hyperintensities. Both zinc toxicity and copper deficiency will lead to low whole-blood copper concentration.
Treatment of zinc toxicity is directed at removing exposure. Chelation has no established role in zinc toxicity. Toxicity from using Zn to treat COVID-19 has not been reported [95].
Ascorbic Acid (Vitamin C)
In the spring of 2020, physicians reported using intravenous vitamin C for those in septic shock from COVID-19 motivated by a clinical trial in 2017 of 47 patients with septic shock and 47 historical controls [96]. In 2021, an observational study of 67 patients admitted to the ICU with ARDS from COVID-19 found that the median plasma vitamin C level was 0.14 mg/dL (interquartile range, 0.10–1.08; reference value 0.2–0.4 mg/dL) but did not control for dietary vitamin C intake nor assess causality by supplementing vitamin C [97]. A multicenter randomized controlled trial in China in 2021 on 56 patients with probable COVID-19 found no difference in 28-day mortality or need for mechanical ventilation between those who received an intravenous infusion of 12 g of vitamin C every 12 h over infusing an equivalent volume of sterile water (100 mL) and reported no serious adverse events [98].
Vitamin C toxicity can manifest as gastrointestinal discomfort, acid reflux, increased frequency of kidney stones, hemolysis, and rectal bleeding. Two patients hospitalized with COVID-19 who received an infusion of 50 mg/kg vitamin C four times a day developed intrarenal oxalate nephropathy and acute tubular necrosis, although before receiving vitamin C they had been in septic shock [99]. As a water-soluble vitamin, the body has a limited ability to store vitamin C. The best treatment is to cease excessive ingestion. Potassium citrate may decrease the frequency of calcium oxalate kidney stones in those who continue to consume excess vitamin C [100].
Colloidal Silver
Colloidal silver refers to silver particles suspended in a solution, usually water, for ingestion. It is a perennial cure-all with no demonstrated health benefit [101, 102]. Prolonged use of silver may lead to permanent bluish-greyish skin discoloration termed argyria. Nanosilver refers to the same formulation with “smaller particles.” Neither formulation is approved by the FDA nor regulated by the FDA for purity or accurate compounding. There are no reports to Poison Control of toxicity from colloidal silver used for COVID-19.
A clinical trial of 30 mL of colloidal silver three times a day for 5 days taken orally or via nebulizer has been registered at ClinicalTrials.gov (NCT04978025) and recruited participants in Tunisia from 2020 to 2021. The FDA has not approved silver for enteral use. Human exposure to nebulized colloidal silver has not been formally reported. The highest concentration of silver vapor rats could be exposed to for 5 days a week for 3 months without develo** any organ impairments was 0.11–0.75 mg/m3 [103]. For comparison, the NOAEL (no observable adverse effect limit) for carbon monoxide is 3 mg/m3 [104]. There is no known treatment for hemodynamic collapse from a colloidal silver overdose.
Conclusion
In the first year of the COVID-19 pandemic, pharmaceutical companies repurposed existing therapeutics against SARS-CoV-2. This led to concerns about toxicity and drug-drug interactions from the use of therapeutics typically used by subspecialists in a wider population and at higher doses than were known to be safe. In the second year, opportunists promoted “remedies” such as ivermectin, colloidal silver, and zinc as safer than vaccines, based on flimsy or fictional evidence. In the third year, the development of targeted antivirals and monoclonal antibodies has provided additional therapies for those who cannot receive or adequately respond to a vaccine. Data from the first 3 years of the pandemic on repurposed medications suggests most are neither as effective as hoped nor toxic as feared.
In opting for alternative remedies, people may choose ineffective treatments over effective treatments, experience drug-drug interactions when they combine treatments, or take toxic amounts of ineffective treatments. To obtain alternative remedies, people may turn to sources that are not regulated enough to guarantee a product that is safe for human consumption. Acquiring medications such as hydroxychloroquine or ivermectin for futile purposes may deprive those who need the medication.
Online misinformation and disinformation have perpetuated the use of ineffective substances. Preprints, whose conclusions are easy to disseminate but methods and results are unvetted, obscure efforts to identify safe and effective therapeutics. The distinction between a preprint that is publicly disseminated and not peer-reviewed and a preprint that has been peer-reviewed but not yet formatted for a journal may not be clear, even to a researcher. It is challenging to correctly integrate the avalanche of computational, animal, cell culture, and human findings even when restricting oneself to peer-reviewed studies. Evidence for the efficacy and safety of a drug at one dose is not evidence of its safety and efficacy at other doses.
References
Boehmer TK, Kompaniyets L, Lavery AM, Hsu J, et al. Association between COVID-19 and myocarditis using hospital-based administrative data - United States, March 2020-January 2021. MMWR Morb Mortal Wkly Rep. 2021;70(35):1228–32.
Chary MA, Barbuto AF, Izadmehr S, Hayes BD, Burns MM. COVID-19: therapeutics and their toxicities. J Med Toxicol. 2020;16(3):284–94.
Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;(3):141–54. https://doi.org/10.1038/s41579-020-00459-7.
Gautret P, Lagier JC, Parola P, Hoang VT, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56(1):105949.
Chen Z, Hu J, Zhang Z, Jiang S, et al. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. https://doi.org/10.1101/2020.03.22.20040758.
Axfors C, Schmitt AM, Janiaud P, Van't Hooft J, et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat Commun. 2021;12(1):2349.
Perez J, Roustit M, Lepelley M, Revol B, et al. Reported adverse drug reactions associated with the use of hydroxychloroquine and chloroquine during the COVID-19 pandemic. Ann Intern Med. 2021;174(6):878–80.
Vicenti I, Zazzi M, Saladini F. SARS-CoV-2 RNA-dependent RNA polymerase as a therapeutic target for COVID-19. Expert Opin Ther Pat. 2021;31(4):325–37.
Kokic G, Hillen HS, Tegunov D, Dienemann C, et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat Commun. 2021;12(1):279.
** Z, Kinkade A, Behera I, Chaudhuri S, et al. Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antiviral Res. 2017;143:151–61. https://doi.org/10.1016/J.ANTIVIRAL.2017.04.005.
Elsawah HK, Elsokary MA, Abdallah MS, ElShafie AH. Efficacy and safety of remdesivir in hospitalized Covid-19 patients: Systematic review and meta-analysis including network meta-analysis. Rev Med Virol. 2021;31(4):e2187.
Spinner CD, Gottlieb RL, Criner GJ, Arribas López JR, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: A randomized clinical trial. JAMA. 2020;324(11):1048–57.
Pettit NN, Pisano J, Nguyen CT, Lew AK, et al. Remdesivir use in the setting of severe renal impairment: a theoretical concern or real risk? Clin Infect Dis. 2021;73(11):e3990–5.
Gérard AO, Laurain A, Fresse A, Parassol N, et al. Remdesivir and acute renal failure: a potential safety signal from disproportionality analysis of the WHO safety database. Clin Pharmacol Ther. 2021;109(4):1021–4.
Luke DR, Tomaszewski K, Damle B, Schlamm HT. Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J Pharm Sci. 2010;99(8):3291–301.
Shah S, Ackley TW, Topal JE. Renal and hepatic toxicity analysis of remdesivir formulations: does what is on the inside really count? Antimicrob Agents Chemother. 2021;65(10):e0104521.
Sanchez-Codez MI, Rodriguez-Gonzalez M, Gutierrez-Rosa I. Severe sinus bradycardia associated with Remdesivir in a child with severe SARS-CoV-2 infection. Eur J Pediatr. 2021;180(5):1627.
Gubitosa JC, Kakar P, Gerula C, Nossa H, et al. Marked Sinus Bradycardia Associated with Remdesivir in COVID-19: A Case and Literature Review. JACC Case Rep. 2020;2(14):2260–4.
Gupta AK, Parker BM, Priyadarshi V, Parker J. Cardiac adverse events with remdesivir in COVID-19 infection. Cureus. 2020;12(10):e11132.
Abdelmajid A, Osman W, Musa H, Elhiday H, et al. Remdesivir therapy causing bradycardia in COVID-19 patients: Two case reports. IDCases. 2021;26:e01254.
Selvaraj V, Bavishi C, Patel S, Dapaah-Afriyie K. Complete heart block associated with remdesivir in COVID-19: a case report. Eur Heart J Case Rep. 2021;5(7):ytab200. https://doi.org/10.1093/ehjcr/ytab200.
Kumar N, Kumar A, Pradhan S, Kumar A, et al. Painful blisters of left hand following extravasation of remdesivir infusion in COVID-19. Indian J Crit Care Med. 2021;25(2):240–1.
van Merendonk LN, Leeuwerik AF, den Brok MWJ, Hekking PW, et al. Peripheral infiltration of remdesivir in 3 patients with COVID-19: Case series and discussion. Am J Health Syst Pharm. 2021;78(21):1944–51.
** Z, Smith LK, Rajwanshi VK, Kim B, et al. The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) Ribofuranosyl 5'-triphosphate towards influenza A virus polymerase. PLoS One. 2013;8(7):e68347.
Hung DT, Ghula S, Aziz JMA, Makram AM, et al. The efficacy and adverse effects of favipiravir on patients with COVID-19: A systematic review and meta-analysis of published clinical trials and observational studies. Int J Infect Dis. 2022;120:217–27.
Ruzhentsova TA, Oseshnyuk RA, Soluyanova TN, Dmitrikova EP, et al. Phase 3 trial of Coronavir (favipiravir) in patients with mild to moderate COVID-19. Am J Transl Res. 2021;13(11):12575–87.
Toyama Chemical Company. Report on approval of favipiravir. 2011 Mar. https://www.pmda.go.jp/files/000210319.pdf. Accessed 14 Jan 2022.
Holman W, Holman W, McIntosh S, Painter W, et al. Accelerated first-in-human clinical trial of EIDD-2801/MK-4482 (molnupiravir), a ribonucleoside analog with potent antiviral activity against SARS-CoV-2. Trials. 2021;22(1):561.
Malone B, Campbell EA. Molnupiravir: coding for catastrophe. Nat Struct Mol Biol. 2021;28(9):706–8.
Sticher ZM, Lu G, Mitchell DG, Marlow J, et al. Analysis of the potential for N4-hydroxycytidine to inhibit mitochondrial replication and function. Antimicrob Agents Chemother. 2020;64(2):e01719.
Marroquin LD, Hynes J, Dykens JA, Jamieson JD, et al. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97(2):539–47.
Merck. Molnupiravir (Mk-4482) FDA advisory committee meeting briefing document. 2021.
Painter WP, Holman W, Bush JA, Almazedi F, et al. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob Agents Chemother. 2021;65(5):e02428–0.
Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, et al. MOVe-OUT Study Group. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. NEJM. 2022;386(6):509–20.
Merck. Important safety information regarding use of molnupiravir in pregnancy and individuals of childbearing potential. 2021. https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html. Accessed 7 Mar 2022.
Lagevrio 200 mg hard capsules - Summary of Product Characteristics (SmPC) - (EMC). Available from https://www.medicines.org.uk/emc/product/13044. Accessed 7 Mar 2022.
Mitsuya H, Maeda K, Das D, Ghosh AK. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv Pharmacol. 2008;56:169–97.
Vangeel L, Chiu W, De Jonghe S, Maes P, et al. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antiviral Res. 2022;198:105252. https://doi.org/10.1016/j.antiviral.2022.105252.
Hull MW, Montaner JS. Ritonavir-boosted protease inhibitors in HIV therapy. Ann Med. 2011;43(5):375–88.
Rock BM, Hengel SM, Rock DA, Wienkers LC, et al. Characterization of ritonavir-mediated inactivation of cytochrome P450 3A4. Mol Pharmacol. 2014;86(6):665–74. https://doi.org/10.1124/mol.114.094862.
Cao B, Wang Y, Wen D, Liu W, et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. 2020;382(19):1787–99. https://doi.org/10.1056/NEJMoa2001282.
Liu F, Xu A, Zhang Y, Xuan W, et al. Patients of COVID-19 may benefit from sustained Lopinavir-combined regimen and the increase of Eosinophil may predict the outcome of COVID-19 progression. Int J Infect Dis. 2020;95:183–91.
Young BE, Ong SWX, Kalimuddin S, Low JG, et al. Singapore 2019 novel coronavirus outbreak research team. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA. 2020;323(15):1488–94.
Lampiris HW, Koss CA. Lopinavir. In: Grayson ML, Cosgrove SE, Crowe S, et al., editors. Kucers' the use of antibiotics: a clinical review of antibacterial, antifungal, antiparasitic, and antiviral drugs. 7th ed: CRC Press; 2017. p. 4087–103.
Florence E, Schrooten W, Verdonck K, Dreezen C, et al. Rheumatological complications associated with the use of indinavir and other protease inhibitors. Ann Rheum Dis. 2002 Jan;61(1):82–4. https://doi.org/10.1136/ard.61.1.82.
Cresswell FV, Tomlins J, Churchill DR, Walker-Bone K, et al. Achilles tendinopathy following Kaletra (lopinavir/ritonavir) use. Int J STD AIDS. 2014;25(11):833–5.
Ahmad B, Batool M, Ain QU, Kim MS, et al. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int J Mol Sci. 2021;22:9124.
Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–93.
LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012–. Paxlovid.
Stork CM. Antibacterials, antifungals, and antivirals. In: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR, editors. Goldfrank's toxicologic emergencies, 10e: McGraw Hill; 2015.
Lange NW, Salerno DM, Jennings DL, Choe J, et al. Nirmatrelvir/ritonavir use: Managing clinically significant drug-drug interactions with transplant immunosuppressants. Am J Transplant. 2022 Jul;22(7):1925–6.
Cvetkovic RS, Goa KL. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs. 2003;63(8):769–802.
Catlin NR, Bowman CJ, Campion SN, Cheung JR, et al. Reproductive and developmental safety of nirmatrelvir (PF-07321332), an oral SARS-CoV-2 Mpro inhibitor in animal models. Reprod Toxicol. 2022 Mar;108:56–61.
RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19—preliminary report. N Engl J Med. Mass Medical Soc. 2020;384:693–704. https://doi.org/10.1056/nejmoa2021436.
RECOVERY Collaborative Group, Horby P, Lim WS, Emberson JR, et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384(8):693–704.
Taper C. Systemic corticosteroid--associated psychiatric adverse effects. US Pharm. 2016;41:16–8.
Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361–7.
Owens RC Jr, Nolin TD. Antimicrobial-associated QT interval prolongation: pointes of interest. Clin Infect Dis. 2006;43(12):1603–11.
RECOVERY Collaborative Group. Azithromycin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397(10274):605–12.
Mercuro NJ, Yen CF, Shim DJ, Maher TR, et al. Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(9):1036–41.
Lagier JC, Million M, Gautret P, Colson P, et al. Outcomes of 3,737 COVID-19 patients treated with hydroxychloroquine/azithromycin and other regimens in Marseille, France: A retrospective analysis. Travel Med Infect Dis. 2020;36:101791.
Tucker ME. ESC says continue hypertension meds despite COVID-19 concern. Medscape. 2020; https://www.medscape.com/viewarticle/926838. Accessed 15 Mar 2022.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020;8(4):e21.
Yang X, Yu Y, Xu J, Shu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–81.
Zhang P, Zhu L, Cai J, Lei F, et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. 2020;126(12):1671–81.
Biswas M, Kali MSK. Association of angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers with risk of mortality, severity or SARS-CoV-2 test positivity in COVID-19 patients: meta-analysis. Sci Rep. 2021;11(1):5012.
Martin RJ, Robertson AP, Choudhary S. Ivermectin: an anthelmintic, an insecticide, and much more. Trends Parasitol. 2021;37(1):48–64.
Griffin J, Fletcher N, Clemence R, Blanchflower S, et al. Selamectin is a potent substrate and inhibitor of human and canine P-glycoprotein. J Vet Pharmacol Ther. 2005;28(3):257–65.
Caly L, Druce JD, Catton MG, Jans DA, et al. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res. 2020;178:104787.
Stokel-Walker C. Ivermectin buyers’ clubs. New Sci. 2021;251(3355):8–9.
FDA Letter to Stakeholders: Do not use ivermectin intended for animals as treatment for COVID-19 in humans. 2022. Available from https://www.fda.gov/animal-veterinary/product-safety-information/fda-letter-stakeholders-do-not-use-ivermectin-intended-animals-treatment-covid-19-humans. Accessed 8 Jan 2022.
Bray M, Rayner C, Noël F, Jans D, et al. Ivermectin and COVID-19: A report in Antiviral Research, widespread interest, an FDA warning, two letters to the editor and the authors’ responses. Antiviral Res. 2020;178:104805.
Bryant A, Lawrie TA, Dowswell T, Fordham EJ, et al. Ivermectin for prevention and treatment of COVID-19 infection: a systematic review, meta-analysis, and trial sequential analysis to inform clinical guidelines. Am J Ther. 2021;28(4):e434–60.
Turkia M. A timeline of ivermectin-related events in the COVID-19 pandemic. 2021. https://doi.org/10.13140/RG.2.2.13705.36966
Hobbs CA, Saigo K, Koyanagi M, Hayashi S. Magnesium stearate, a widely used food additive, exhibits a lack of in vitro and in vivo genotoxic potential. Toxicol Rep. 2017;16(4):554–9.
Williams GM, Iatropoulos MJ, Whysner J. Safety assessment of butylated hydroxyanisole and butylated hydroxytoluene as antioxidant food additives. Food Chem Toxicol. 1999;37(9-10):1027–38.
Package insert for STROMECTOL ® (IVERMECTIN). Merck. 2007. https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/050742s026lbl.pdf. Accessed 24 Jun 2022.
Guzzo CA, Furtek CI, Porras AG, Chen C, et al. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J Clin Pharmacol. 2002;42(10):1122–33.
Mayer TW, Horton ML. Modulation of monomethylhydrazine-induced seizures by ivermectin. Toxicol Lett. 1991;57(2):167–73.
Abd-Elsalam S, Noor RA, Badawi R, Khalaf M, et al. Clinical study evaluating the efficacy of ivermectin in COVID-19 treatment: A randomized controlled study. J Med Virol. 2021;93(10):5833–8.
Rajter JC, Sherman MS, Fatteh N, Vogel F, et al. Use of ivermectin is associated with lower mortality in hospitalized patients with coronavirus disease 2019: The Ivermectin in COVID nineteen study. Chest. 2021;159(1):85–92.
Temple C, Hoang R, Hendrickson RG. toxic effects from ivermectin use associated with prevention and treatment of Covid-19. N Engl J Med. 2021;385(23):2197–8.
Nakae Y, Kanaya N, Namiki A. The direct effects of diazepam and midazolam on myocardial depression in cultured rat ventricular myocytes. Anesth Analg. 1997;85(4):729–33.
Follmer CH, Lum BK. Protective action of diazepam and of sympathomimetic amines against amitryptyline-induced toxicity. J Pharmacol Exp Ther. Citeseer. 1982;222:424–9.
Atack JR, Scott-Stevens P, Beech JS, Fryer TD, et al. Comparison of lorazepam [7-chloro-5-(2-chlorophenyl)-1,3-dihydro-3-hydroxy-2H-1,4-benzodiazepin-2-one] occupancy of rat brain gamma-aminobutyric acid(A) receptors measured using in vivo [3H]flumazenil (8-fluoro 5,6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid ethyl ester) binding and [11C]flumazenil micro-positron emission tomography. J Pharmacol Exp Ther.
Elgarf AA, Siebert DCB, Steudle F, Draxler A, et al. Different benzodiazepines bind with distinct binding modes to GABA A receptors. ACS Chem Biol. 2018;13(8):2033–9.
Estrada-Mondragon A, Lynch JW. Functional characterization of ivermectin binding sites in α1β2γ2L GABA(A) receptors. Front Mol Neurosci. 2015 Sep;25(8):55.
Chiang KC, Gupta A. To zinc or not to zinc for COVID-19 prophylaxis or treatment? J Med Microbiol. 2021;70(9).
The Silver Bullet Against Coronavirus is HERE! Imgflip. 2022. https://archive.ph/CMCVm. 6 Jan 2022.
Did a Noted Pathologist Write This Viral Coronavirus Advice Letter? Snopes.com. Available from https://www.snopes.com/fact-check/zinc-lozenges-coronavirus/. Accessed 6 Jan 2022.
Maggini S, Beveridge S, Suter M. A combination of high-dose vitamin c plus zinc for the common cold. J Int Med Res. 2012;40(1):28–42.
Thomas S, Patel D, Bittel B, Wolski K, et al. Effect of high-dose zinc and ascorbic acid supplementation vs usual care on symptom length and reduction among ambulatory patients with SARS-CoV-2 infection: The COVID A to Z randomized Clinical Trial. JAMA Netw Open. 2021;4(2):e210369.
Saper RB, Rash R. Zinc: An Essential Micronutrient. Am Fam Physician. 2009;79(9):768–72.
Francis Z, Book G, Litvin C, Kalivas B. The COVID-19 Pandemic & Zinc-Induced Copper Deficiency an Important Link. Am J Med. 2022;135(8):e290–1.
Pal A, Squitti R, Picozza M, Pawar A, et al. Zinc and COVID-19: basis of current clinical trials. Biol Trace Elem Res. 2021;199(8):2882–92.
New York hospitals are using vitamin C to treat some coronavirus patients. Newsweek. 2022. https://www.newsweek.com/new-york-hospitals-vitamin-c-coronavirus-patients-1494407. Accessed 12 Jan 2022.
Tomasa-Irriguible TM, Bielsa-Berrocal L. COVID-19: Up to 82% critically ill patients had low Vitamin C values. Nutr J. 2021;20(1):66.
Zhang J, Rao X, Li Y, Zhu Y, et al. Pilot trial of high-dose vitamin C in critically ill COVID-19 patients. Ann Intensive Care. 2021;11(1):5.
Fontana F, Cazzato S, Giovanella S, Ballestri M, et al. Oxalate nephropathy caused by excessive vitamin C administration in 2 patients with COVID-19. Kidney Int Rep. 2020;5(10):1815–22.
Frassetto L, Kohlstadt I. Treatment and prevention of kidney stones: an update. Am Fam Physician. 2011;84(11):1234–42.
Medici S, Peana M, Nurchi VM, Zoroddu MA. Medical uses of silver: history, myths, and scientific evidence. J Med Chem. 2019;62(13):5923–43.
Lansdown ABG. Critical observations on the neurotoxicity of silver. Crit Rev Toxicol. 2007;37(3):237–50.
Hadrup N, Sharma AK, Loeschner K, Jacobsen NR. Pulmonary toxicity of silver vapours, nanoparticles and fine dusts: A review. Regul Toxicol Pharmacol. 2020;115:104690.
ATSDR. Toxicological profile for carbon monoxide. 2010. p. 1–347. https://www.atsdr.cdc.gov/toxprofiles/tp201.pdf. Accessed 14 Jan 2022.
Acknowledgements
We thank Michael Iocco for useful discussions and edifying comments.
Funding
Dr. Izadmehr is supported by the Loan Repayment Program (National Center for Advancing Translational Sciences, National Institutes of Health) and the T32 Cancer Biology Training Program (5T32CA078207, National Cancer Institute, National Institutes of Health). Dr. Chary is supported by the Loan Repayment Program (National Institute for Drug Abuse, National Institutes of Health).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
There are no Conflicts of Interest.
Additional information
Supervising Editor: Michael Hodgman, MD
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chary, M.A., Barbuto, A.F., Izadmehr, S. et al. COVID-19 Therapeutics: Use, Mechanism of Action, and Toxicity (Xenobiotics). J. Med. Toxicol. 19, 26–36 (2023). https://doi.org/10.1007/s13181-022-00918-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13181-022-00918-y