Search
Search Results
-
The FMO-DFTB Method
Although the fragment molecular orbital (FMO) method enables electronic structure calculations with near-linear scaling behavior with respect to...
-
Reliability of semiempirical and DFTB methods for the global optimization of the structures of nanoclusters
In this work, we explore the possibility of using computationally inexpensive electronic structure methods, such as semiempirical and DFTB...

-
Performance study of the electronic and optical parameters of thermally activated delayed fluorescence nanosized emitters (CCX-I and CCX-II) via DFT, SCC-DFTB and B97-3c approaches
Conventional density functional theory (DFT) is the common choice of researchers. However, recently the density functional tight binding (DFTB)...

-
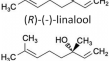
Quantum chemical study on inclusion of linalool into cucurbiturils
Inclusion of one of the natural enantiomers of linalool ((R)-(-)-linalool, simply LIN) into cucurbit[ n ]urils (CB[ n ]s) was investigated by means of...
-
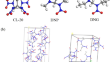
Thermal decomposition mechanisms of energetic CL-20-based co-crystals: quantum molecular dynamics simulations
The decomposition mechanisms of energetic CL-20:2,4-dinitro-2,4-diazapentane (DNP) and CL-20:2,4-dinitro-2,4-diazaheptane (DNG) co-crystals at high...
-
Theoretical studies on structure and dynamics of anatase TiO2 (101)/H2SO4/H2O interface in the early stage of titania sulfation
This work provides a molecular-level insight into the structural and dynamical processes of H 2 SO 4 /H 2 O molecules on anatase TiO 2 (101) surface using...

-
Ground-to-excited derivative couplings for the density functional-based tight-binding method: semi-local and long-range corrected formulations
A derivation of non-adiabatic coupling vectors for the density functional-based tight-binding method (DFTB) between ground and excited states is...

-
Exploring energy landscapes at the DFTB quantum level using the threshold algorithm: the case of the anionic metal cluster Au\(_{20}^{-}\)
AbstractWe report the combination of the threshold algorithm with the Density Functional-based Tight Binding method allowing for the exploration of...

-
The structure of 1,3-butadiene clusters
Molecular clusters of 1,3-butadiene were theoretically investigated using a variety of approaches, encompassing classical force fields and different...

-
Inclusion of thymol into cucurbiturils: density functional theory approach with dispersion correction and natural bond orbital analysis
Stability of inclusion complexes of thymol (a natural flavour) with cucurbit[ n ]urils was interpreted by using density functional theory with...

-
Effect of Orientational Isomerism in Neutral Water Hexamers on Their Thermodynamic Properties and Concentrations in the Gas Phase
In order to estimate the effect of hydrogen bond network isomerism on the thermodynamic functions and concentrations of water clusters in the gas...

-
A new active learning approach for adsorbate–substrate structural elucidation in silico
Adsorbate interactions with substrates (e.g. surfaces and nanoparticles) are fundamental for several technologies, such as functional materials,...

-
Interaction between chloride ions mediated by carbon nanotubes: a chemical attraction
The interaction between two Cl − ions separated by the wall of a narrow carbon nanotube has been investigated by density functional theory (DFT) and...

-
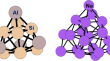
A new active learning approach for global optimization of atomic clusters
In catalysis, an accurate structural elucidation of molecules, atomic clusters, nanoparticles and solid surfaces is required to understand chemical...
-
Molecular Dynamics Simulations and Intermolecular Forces
In this chapter, the main computational methods presently used in molecular modelling to compute the energy of an assembly of molecules and to...
-
Theoretical studies of size effects on surfacial properties for CL-20 and NTO nanoparticles
Nanoparticles (NPs) possess the unique physical and chemical properties compared with gas-phase molecule and bulk crystal. Hence, the energetics,...

-
Theorectical Studies on Surface-Induced Energetic, Electronic, and Vibrational Properties of Triamino-trinitrobenzene Nanoparticles
The energetics, electronic features, and vibrational properties of a series of triamino-trinitrobenzene (TATB) nanoparticles (NPs) were investigated...

-
The FP-LAPW/GAM-MPW1K approach: a reliable abinitio method for calculating the band gap of II-VI semiconductors monochalcogenides
ContextThe bandgap of metal monochalcogenides (MMCs) is a key property that governs their physical and chemical properties. Accurate measurement of...
-
Exciton effect in new generation of carbon nanotubes: graphdiyne nanotubes
Graphdiyne-based nanotubes (GDNTs) are a novel type of carbon nanotubes. While conventional carbon nanotubes (CNTs) are generated by rolling graphene...

-
Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential
Atomistic simulation has a broad range of applications from drug design to materials discovery. Machine learning interatomic potentials (MLIPs) have...
