
Abstract
Background
Bigfin squid is one of the economically important seafood resources in Vietnam’s fisheries and the waters around Con Dao and Phu Quoc islands are two major fishing grounds where this species has been actively exploited. The start codon targeted polymorphism (SCoT) and CAAT box–derived polymorphism (CBDP) techniques were used to generate DNA fingerprinting data to analyze the genetic diversity, variation, and structure of the two populations in the waters surrounding Phu Quoc and Con Dao islands together with mitochondrial cytochrome C oxidase subunit I (COI) gene sequence data.
Results
Con Dao population possessed a higher diversity [expected heterozygosity (He) = 0.2254, Shannon index (I) = 0.3459, percentage of polymorphic bands (PPB) = 80.14%, nucleotide diversity (π) = 0.0336, haplotype diversity (h) = 0.910 with 16 haplotypes] than Phu Quoc population (He = 0.1854, I = 0.2873, PPB = 70.38%, π = 0.0246, h = 0.838 with 14 haplotypes). The genetic diversity at species level in the investigated region was at level of He = 0.2169, I = 0.3399, PPB = 86.41, π = 0.0289, and h = 0.892 with 24 haplotypes. Based on DNA fingerprinting data, the pairwise genetic similarity coefficients among individuals of the Con Dao population were lower (average of 0.7977) than the Phu Quoc population (average of 0.8316). Based on mitochondrial COI data, the pairwise genetic distances among individuals of the Con Dao population were higher (average of 0.0361) than the Phu Quoc population (average of 0.0263). Gene differentiation (GST) between two investigated populations was 0.0316 and 0.0310 leading to the genetic distance was 0.0573 and 0.0213 and the gene flow between them was Nm = 8.2209 and 11.4700 migrants per generation among populations based on DNA fingerprinting and based on COI gene sequence data, respectively. Genetic variation within individuals of both populations (WP) played the key role in total genetic variation at species level in surveyed region.
Conclusions
For the bigfin reef squid species in the surveyed region, the Con Dao population had the higher genetic diversity than the Phu Quoc population, between them existed a low to moderate genetic differentiation and a genetic exchange via gene flow. The DNA fingerprinting data better revealed the genetic differentiation between the two surveyed populations while the mitochondrial COI gene sequence data could show the phylogenetic relationship among the surveyed individuals and the other from the sea regions in Southeast Asia. Based on the results obtained, fisheries management strategies are suggested toward the conservation and sustainable exploitation of this species.
Similar content being viewed by others

Background
The bigfin reef squid, scientifically known as Sepioteuthis lessoniana d’Orbigny, is one of the members of the Loliginidae family. This species is considered to be the most widely distributed loliginid squid in the Indo-West Pacific region because besides its largest distribution range from Japan to northern Australia, it also occurs in many other places in the Pacific and Indian Oceans such as New Zealand, the Hawaiian Islands, South India, West Africa, the Madagascar island, Red Sea, and Western Mediterranean [1]. In Vietnam, bigfin reef squid occurs in the waters of the Gulf of Tonkin, the Southern Central, the Southern East, and the Southern West [2,3,4] and has been considered as one of the most economically important seafood resources.
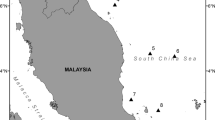
In its distribution areas, this species usually occurs in water from the surface to a depth of about 100 m, in coastal habitats with the bottom layer usually covered by seagrass beds and coral reefs [1]. Such habitats are relatively common in the Southwestern and Southeastern seas of Vietnam, which are characterized by the coastal habitats of the two large islands, Phu Quoc and Con Dao. In fact, bigfin reef squid used to be fairly abundant and has been considered as a famous seafood in these two islands compared to other distribution areas in Vietnam. These two islands are separated by about 345 km as the crow flies and the shortest sea distance between them is about 410 km. Sharing characteristics with other loliginid squids, the planktonic paralarvae of S. lessoniana are widely distributed after hatching by coastal and ocean currents [5, 6] while juveniles and adults live in coastal and consistently move inshore to start mating and spawning [1]. These indicated that adult squid individuals from Con Dao and from Phu Quoc are almost impossible to mate together directly, and therefore bigfin reef squid individuals from Con Dao island and from Phu Quoc island must belong to two separate populations.
Bigfin reef squid in particular and squid in general are crucial components in the food chains and webs as well as the biodiversity of marine ecosystems and are seafood of high economic value for fisheries. Despite playing such important role, the species S. lessoniana has faced to two risks, including overexploitation and loss of suitable habitat as a result of climate change [7, 8] which can lead to species extinction and loss of valuable marine resources without effective strategies for conservation and sustainable exploitation.
Such strategies are often established based on a fundamental conception of the importance of intraspecific genetic diversity, which is determined by the genetic diversity of the constituent populations and the gene flow among them and is measured at markers scattered throughout the genome [9]. Low population genetic diversity is often thought to be a consequence of inbreeding degradation and increased genetic drift and this low diversity itself is the cause to reduce individual vitality, along with a depleted capacity for population growth [10]. On the other hand, high genetic diversity is often considered as the driving force to promote population survival and ensure the ability of populations to adapt to changing environmental pressures [11].
In case of the bigfin reef squid in Vietnam, there were several populations which used to be evaluated for genetic diversity, including the population in the Gulf of Tonkin based on the mitochondrial DNA noncoding region [3] and populations in Nha Trang Bay and Phu Quoc island based on mitochondrial COI and 16S rRNA sequences [4]. In the study by Cheng et al. in 2013, the haplotype diversity and the nucleotide diversity were respectively determined at 0.73 and 0.004 for the S. lessoniana population in Phu Quoc; however, this data was only based on 17 samples. Thus, population genetic data of bigfin reef squid in Con Dao and Phu Quoc islands have been still lacking and need to be supplemented to create the scientific basis for establishment of conservation and exploitation strategies [4].
For bigfin reef squid, previous studies on population genetic diversity were conducted by two groups of methods, including using mitochondrial DNA sequence data [3, 4] and using DNA fingerprinting data generated through the allozyme marker [12] and microsatellite marker [13, 14].
In general, DNA fingerprinting data can be generated using many markers such as simple sequence repeat (SSR), inter simple sequence repeat (ISSR) [15, 16], random amplified polymorphic DNA (RAPD), and amplified fragment length polymorphism (AFLP) [17]. Although not direct reflecting the allele status of the locus, dominant markers still have been widely used in population genetic diversity assessment due to their high sensitivity in expressing the differences among the surveyed individuals. With the development of molecular biology, many dominant markers have been developed recently, such as start codon targeted polymorphism (SCoT) [18] and CAAT box–derived polymorphism (CBDP) [19] markers. SCoT and CBDP markers target the start codon and CAAT box of functional genes, respectively. Both of these markers are widely used in studies of population genetics in plants; while SCoT markers have also been used in studies of population genetics in animals [20,21,22], there are almost no found publications on the use of CBDP markers in this field. However, in the gene structure in eukaryotes, the CAAT box is located upstream of the transcription initiation site and signals the binding site for the RNA transcription factor, namely, NF-Y subunits; the sequence of 5′-GGTTA-3′ is conserved for the CAAT box [23].
With the aim of determining the genetic diversity and variation of the two bigfin reef squid populations in Con Dao and Phu Quoc islands and the genetic differentiation between them, in this study, in addition to mitochondrial COI gene sequence data, DNA fingerprinting data generated using two techniques of SCoT and CBDP were also used for analysis.
Methods
Materials
Samples were collected in October 2020 by night fishing with lure-hooks in the same way to exploit form local fishermen. In a total of collected 59 juvenile and adult samples of S. lessoniana, 28 samples belonged to the Con Dao population were caught in the waters around Con Dao island, and 31 samples belonged to the Phu Quoc population were caught in the waters around Phu Quoc island. Fishing localities of these samples were not more than 10 km offshore. Collected specimens were tentacular tissues of captured individuals, individually labeled and preserved in 95% ethanol at −20 °C until DNA extraction. The field trip sample notation was done continuously for all the caught samples, but only samples belonging to the S. lessoniana species were used in this study.
Twenty-eight representative samples of the Con Dao population were assigned as Sles036, Sles037, Sles039–Sles049, Sles052–Sles055, Sles057, Sles058, Sles060–Sles065, Sles067, Sles068, and Sles070. Thirty-one representative samples of the Phu Quoc population were assigned as Sles072–Sles091 and Sles093–Sles103. Samples were not numbered consecutively as above due to exclusion of different species individuals during fishing.
DNA extraction
Total genomic DNA was extracted from ethanol-preserved tentacular tissue using cetyltrimethylammonium bromide (CTAB) method [24] which modified by Adamkewicz and Harasewych [25]. Three DNA samples were extracted from each individual sample. The DNA concentration and quality were measured using spectrophotometry method [26] using a NanoScan2 system (Analytik Jena). The DNA samples with OD260/OD280 values between 1.8 and 2.0 were kept at −20 °C for the subsequent PCRs.
DNA fingerprinting
In this study, DNA fingerprinting data was obtained by using SCoT and CBDP techniques. PCRs were performed in 50 μL reactions containing 25 μL My Red HS Taq mix (Bioline), 0.2 μM primer, and approximately 30 ng DNA templates. The PCRs were performed using an Eppendorf Mastercycler Pro S thermal cycler (Germany). The thermal program used in SCoT technique was as follows: initial denaturation at 94 °C for 5 min; 36 cycles of 94 °C for 15 s, 50 °C for 15 s, 72 °C for 45 s; final extension at 72 °C for 10 min [18]. The thermal program used in CBDP technique: initial denaturation at 94 °C for 5 min; 6 cycles of 94 °C for 45 s, 35 °C for 45 s, 72 °C for 90 s; 30 cycles of 94 °C for 45 s, 51 °C for 45 s, 72 °C for 90 s; final extension at 72 °C for 10 min [19].
After screening, 10 primers for each technique were chosen for PCR; the sequences and amplification features of these primers are shown in Table 1.
To obtain DNA fingerprints for investigated samples, PCR products were separated in 2% agarose gel, electrophoresis was performed at 60 V for 3 h using TBE buffer, the gel then was stained with ethidium bromide (0.5 μg/mL), and photographed under 254/312 nm wavelength lights using UVP GelStudio Plus System (Analytik Jena, Germany).
COI amplification and sequencing
The DNA sequence of the mitochondrial COI gene was isolated amplified using the primer pairs LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) [27]. PCRs were performed in 60 μL reactions containing 30 μL My Red HS Taq mix (Bioline), 0.4 μM each primer, and approximately 40 ng DNA templates using the Eppendorf Mastercycler Pro S thermal cycler (Germany). The thermal program used to amplify COI gene was as follows: initial denaturation at 95 °C for 5 min; 30 cycles of 95 °C for 15 s, 46 °C for 15 s, 72 °C for 30 s; final extension at 72 °C for 10 min. This program was modified from Folmer et al. [27] and Kim et al. [28] by gradient testing for primer annealing temperature.
PCR products were confirmed by electrophoresis on 1.0% agarose gel electrophoresis and then were purified using AccuPrep® Gel Purification Kits (Bioneer, Korea). Purified DNA samples together with two primers LCO1490 and HCO2198 were preserved in icepack during sending to 1st BASE DNA Sequencing Services (Singapore) for sequencing. DNA sequencing was performed by Sanger method using ABI 3730 XL sequencer.
Data analysis
The parameters of genetic diversity and variation were calculated for each of two populations and for the whole species in the surveyed region. DNA fingerprint data and mitochondrial COI sequence data were analyzed using different specialized software to calculate the diversity and variation parameters and construct the dendrograms for genetic relationship among studied samples.
Based on DNA fingerprinting data
Since both CBDP and SCoT markers are dominant, each observed band was assumed to represent the genotype at a single biallelic locus [29] and DNA fingerprinting data from them can be combined together for genetic analysis (SCoT and CBDP data sets possessed the same overall utility when being applied in samples of whole species in surveyed region, which were analyzed using iMEC—an online marker efficiency calculator, data not shown). The bands were scored as presence (1) or absence (0) characters to construct the binary data matrix. In studies on population genetic diversity and variation, it is generally recognized that increasing the number of investigated loci gives more reliable results [30]. Accordingly, in this study, the analysis was performed with all bands generated using both SCoT and CBDP techniques.
POPGENE 32 software was used to calculate genetic diversity and variation parameters: the percentage of polymorphic bands (PPB), the expected heterozygosity (He), Shannon index (I), the gene differentiation (GST), the genetic distance between investigated populations (D), and gene flow between them (Nm) [38].
DNA fingerprinting data, which showed close grou** of individuals by population, again better reflects the genetic differentiation and the genetic variation between the two populations compared to mitochondrial COI sequence data. This could be explained by the fact that the SCoT and CBDP data reflect the genetic variation of many genes across different regions of the genome while the mitochondrial COI sequence data only reflect the genetic variation of a single gene.
Analysis of molecular variance (AMOVA) revealed high genetic variation within populations and low genetic differentiation among two populations in investigated regions. Based on the mitochondrial DNA non-coding region, Aoki et al. showed that pairwise gene differentiations among 7 bigfin reef squid populations in Japan, Taiwan, and Gulf of Tonkin of Vietnam were in range of 0.0041 to 0.8593 with the average of 0.3695 [3]. Comparing to this study and the interpretation of gene differentiation values in previous studies [39, 40], the gene differentiation between Con Dao and Phu Quoc populations based on DNA fingerprinting data was at moderate level and based on mitochondrial COI sequence data was at relatively low level.
Based on DNA fingerprinting data, the genetic distance between Con Dao and Phu Quoc populations in current study (D = 0.0361) was significantly higher than between Nagasaki and Rayon populations (D = 0.003, using allozyme technique) [12], although the geographical distance between the Nagasaki and Rayon populations is much farther than between the Con Dao and Phu Quoc populations.
Using mitochondrial COI sequence to investigate 11 populations of bigfin reef squid in Southeast Asia, Cheng et al. showed that the significantly different genetic variation distribution among populations (77.51% or 23.24%) and within populations (22.49% or 76.76%) depended on the investigate lineages [4]. Compared with the results from this study, genetic variation within populations in current study was much higher. This may due to in the presentative sample sets for Con Dao and Phu Quoc populations included different lineages of bigfin reef squid which inherently identified by Cheng et al. and the surveyed region in current study was also smaller.
As mentioned above, the information on the genetic diversity, differentiation, and gene flow among the populations is necessary to establish the strategies for conservation and sustainable exploitation of the species. The findings from the current study indicate that the Con Dao population had a higher genetic diversity than the Phu Quoc population. The main reason for that difference may be that Con Dao island is exposed to strong ocean currents from two different directions, while there has been only a weak ocean current bringing planktonic paralarvae to Phu Quoc island. Accordingly, to maintain the long-term genetic diversity of the populations in surveyed regions, several fishery management strategies including the promotion of programs of artificial propagation and local as well as translocational releasing to increase the size of local populations and to overcome barriers to gene flows among populations; establishing regulations to limit fishing in the breeding season; and enhancing the protection and restoration of coral reefs and seagrass beds to ensure the quality of such habitats, especially at spawning grounds for the species should be fully considered.
Conclusions
For the bigfin reef squid species in the surveyed region, the Con Dao population had the higher genetic diversity than the Phu Quoc population; however, the levels of genetic diversity based on COI data of these populations were at relatively higher compared to other populations in Southeast Asia. Between the investigated populations existed a low to moderate genetic differentiation and a genetic exchange via gene flow.
DNA fingerprinting data better showed the gene differentiation between the two investigated populations than the mitochondrial COI gene sequence data; the COI gene sequence data reflects the phylogenetic relationships of the individuals investigated in the current study with bigfin reef squid in some other sea areas in Southeast Asia.
From this study, several fishery management strategies for conservation and sustainable exploitation of bigfin reef squid are proposed.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its supplementary information files.
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- AP :
-
Distribution of genetic variation among populations
- CBDP:
-
CAAT box–derived polymorphism
- COI:
-
Cytochrome C oxidase subunit 1
- CTAB:
-
Cetyltrimethylammonium bromide
- D :
-
Genetic distance between investigated populations
- G ST :
-
Gene differentiation
- h :
-
Haplotype diversity
- H e :
-
Expected heterozygosity
- I :
-
Shannon index
- NF-Y:
-
Nuclear factor Y
- Nm:
-
Gene flow
- OD260 :
-
Optical density at 260 nm
- OD280 :
-
Optical density at 280 nm
- PCR:
-
Polymerase chain reaction
- PPB :
-
Percentage of polymorphic bands
- SCoT:
-
Start codon targeted polymorphism
- TBE:
-
Tris-borate-EDTA
- UPGMA:
-
Unweighted pair group method with arithmetic mean
- WP :
-
Distribution of genetic variation within populations
- π :
-
Nucleotide diversity
References
Jereb P, Vecchione M, Roper CFE (2010) Family Loliginidae. In: Jereb P, Roper CFE (eds) Cephalopods of the world. An annotated and illustrated catalogue of species known to date. Volume 2. Myopsid and oegopsid squids. FAO species catalogue for fishery purposes, vol 2. FAO, Rome, pp 38–117
Duc NX, Duong TM, Thuy LD (1983) Species composition and exploited production of squid resources in the South Vietnam sea. J Biol 1(2)
Aoki M, Imai H, Naruse T, Ikeda Y (2008) Low genetic diversity of oval squid, Sepioteuthis cf. lessoniana (Cephalopoda: Loliginidae), in Japanese waters inferred from a mitochondrial DNA non-coding region. Pacific Sci 62:403–411
Cheng SH, Anderson FE, Bergman AM, Mahardika GN, Muchlisin ZA, Dang BT, Calumpong HP, Mohamed KS, Sasikumar G, Venkatesan V, Barber PH (2013) Molecular evidence for co-occurring cryptic lineages within the Sepioteuthis cf. lessoniana species complex in the Indian and Indo-West Pacific Oceans. Hydrobiologia 725(1):165–188
Young RE, Harman RF (1988) “Larva”, “paralarva” and “subadult” in cephalopod terminology. Malacologia 29:201–208
Zeidberg L, Hamner W (2002) Distribution of squid paralarvae, Loligo opalescens (Cephalopoda: Myopsida), in the Southern California Bight in the three years following the 1997-1998 El Niño. Marine Biol. 141:111–122. https://doi.org/10.1007/s00227-002-0813-x
Pecl GT, Jackson GD (2007) The potential impacts of climate change on inshore squid: biology, ecology and fisheries. Rev Fish Biol Fish 18(4):373–385
Edwards AJ (2020) Impact of climatic change on coral reefs, mangroves, and tropical seagrass ecosystems. In: Eisma D (ed) Climate change: impact on coastal habitation. Lewis Publishers, Boca Raton, pp 209–234
Teixeira JC, Huber CD (2021) The inflated significance of neutral genetic diversity in conservation genetics. Proc Natl Acad Sci USA 118(10)
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci USA 101:15261–15264
Markert JA, Champlin DM, Gutjahr-Gobell R, Grear JS, Kuhn A, McGreevy TJ, Roth A, Bagley MJ, Nacci DE (2010) Population genetic diversity and fitness in multiple environments. BMC Evol Biol 10(1). https://doi.org/10.1186/1471-2148-10-205
Pratoomchat B, Natsukari Y, Miki I, Chalermwat K (2001) Allozyme determination of genetic diversity in Japanese and Thai populations of oval squid (Sepioteuthis lessoniana Lesson, 1830). La mer 39:133–139
Tomano S, Ahmad-Syazni K, Ueta Y, Ohara K, Umino T (2013) Eleven novel polymorphic microsatellite loci for oval squid Sepioteuthis Lessoniana (shiro-ika type). Int J Mol 14:19971–19975
Zheng XD, Zuo ZR, Su XJ, Li Q (2012) Isolation and characterization of 24 polymorphic microsatellite loci in Sepioteuthis lessoniana. Genet Mol Res 11(4):3961–3965
Serra IA, Procaccini G, Intrieri MC, Migliaccio M, Mazzuca S, Innocenti AM (2007) Comparison of ISSR and SSR markers for analysis of genetic diversity in the seagrass Posidonia oceanica. Mar Ecol Prog Ser 338:71–79
Trieu LN, Bich TT, Ket NV (2022) Population genetic diversity of Camellia Dilinhensis on the Di Linh plateau of Vietnam revealed by ISSR and SCoT markers. Dalat Univ J Sci 12(3):43–55
Borowsky RL (2001) Estimating nucleotide diversity from random amplified polymorphic DNA and amplified fragment length polymorphism data. Mol Phylogenet Evol 18(1):143–148
Collard BCY, Mackill DJ (2008) Start codon targeted (SCoT) polymorphism: a simple, novel DNA marker technique for generating gene-targeted markers in plants. Plant Mol Biol Rep 27(1):86–93
Singh AK, Rana MK, Singh S, Kumar S, Kumar R, Singh R (2013) CAAT box-derived polymorphism (CBDP): a novel promoter-targeted molecular marker for plants. J Plant Biochem Biotechnol 23(2):175–183
Zeinab AM, Mohammad A (2017) Using start codon targeted (SCoT) polymorphism for genetic diversity analysis of three Red Sea fishes (family: Carangidae). IOSR J Pharm Biol Sci 12(1):50–56
Ali HAA (2020) Potential of start codon targeted (SCoT) markers and SDS-PAGE to estimate genetic diversity and relationships among three gastropods species from the Mediterranean Sea, Port Said, Egypt. Egypt J Aquat Biol Fish 24(7):133–143
Ali HAA, Hala EA, Osama AA, Mohamed K (2020) Genetic relationship between two species of genus Dicentrarchus based on SCoT markers and SDS–PAGE. Egypt J Aquat Biol Fish 24(7):393–402
Romier C, Cocchiarella F, Mantovani R, Moras D (2002) The NF-YB/NF-YC structure gives insight into DNA binding and transcription regulation by CCAAT factor NF-Y. J Biol Chem 278(2):1336–1345. https://doi.org/10.1074/jbc.m209635200
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Adamkewicz SL, Harasewych MG (1996) Systematics and biogeography of the genus Donax (Bivalvia: Donacidae) in eastern North America. Am Malacol Bull 13(1):97–103
Weising K, Nybom H, Wolff K, Kahl G (2005) DNA fingerprinting in plants: principles, methods, and applications, (2nd ed.) edn. CRCPress, Taylor & Francis Group. https://doi.org/10.1201/9781420040043
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit 1 from diverse metazoan invertebrates. Mol Marine Biol Biotechnol 3:294–299
Kim DW, Yoo WG, Park HC, Yoo HS, Kang DW, ** SD, Min HK, Paek WK, Lim J (2012) DNA barcoding of fish, insects, and shellfish in Korea. Genom Inform 10:206–211
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535
Carling MD, Brumfield RT (2007) Gene sampling strategies for multi-locus population estimates of genetic diversity. PLoS ONE 2:e160
Yeh FC, Yang R, Boyle TJ, Ye Z, **yan JM (2000) PopGene32, Microsoft Windows-based freeware for population genetic analysis, version 1.32. Molecular Biology and Biotechnology Centre: University of Alberta, Edmonton, Canada
Rohlf FJ (2004) NTSYSpc numerical taxonomy and multivariate analysis system version 2.1 - user guide. Applied Biostatistics Inc.
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28:2537–2539
Excoffier L, Lischer HEL (2010) Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Rozas J, Ferrer-Mata A, JC S-DB, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol 39:783–791
Mingting L, Jun W, Dongxiao W, Arnold LG, Song Y, Paola MR, Guoqing J (2019) Exploring the importance of the Mindoro-Sibutu pathway to the upper-layer circulation of the South China Sea and the Indonesian throughflow. JGR Oceans 124(7):5054–5066. https://doi.org/10.1029/2018JC014910
de Vicente MC, López C, Fulton T (2004) Genetic diversity analysis with molecular marker data: learning module. International Plant Genetic Resources Institute (IPGRI), Rome, Italy
Joana RF (2005) Molecular ecology. John Wiley & Sons Ltd., The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England
Acknowledgements
We would like to thank the Ministry of Science and Technology of Vietnam and Vietnam Academy of Science and Technology for funding, and thank the Dalat University and the Institute of Oceanography, Vietnam, for providing experimental equipment and facilitating this work.
Funding
This study was developed within the framework of the project titled “Studies on dispersal mechanisms of larvae and population connectivity of target marine resources for improving management effectiveness of marine protected areas in the coastal waters from Quang Tri to Kien Giang” under support from the Ministry of Science and Technology of Vietnam (Project code: KC.09.41/16-20), and the project titled “Marine biodiversity in the emerging and submerged banks, offshore islands, and deep-sea waters in South Vietnam” under support from Vietnam Academy of Science and Technology—VAST (Project code: QTRU02.03/23-24).
Author information
Authors and Affiliations
Contributions
LNT conceptualized and supervised the experiment, analyzed data, and was a major contributor in writing the manuscript and was the corresponding author. TTB and NVK conducted the experiment, collected and analyzed data, and wrote the original draft. NVL conceptualized, collected the animal materials and revised the manuscript. All the authors read and agreed to publish the version of manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal samples collected to conduct this study were taken from dead caught individuals as seafood; these individuals belonged to the species that has been allowed to be caught in Vietnam and around the world.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trieu, L.N., Bich, T.T., Van Ket, N. et al. Genetic diversity, variation, and structure of two populations of bigfin reef squid (Sepioteuthis lessoniana d’Orbigny) in Con Dao and Phu Quoc islands, Vietnam. J Genet Eng Biotechnol 21, 116 (2023). https://doi.org/10.1186/s43141-023-00573-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00573-y