
Abstract
Background
Lack of biomarkers and in vitro models has contributed to inadequate understanding of the mechanisms underlying the inferior clinical response to immune checkpoint inhibitors (ICIs) in patients with oncogene-driven non-small cell lung cancer (NSCLC).
Methods
The effect of small molecule tyrosine kinase inhibitors (TKIs) on peripheral blood mononuclear cells (PBMCs) in 34 patients with oncogene-driven NSCLC (cohort A) was compared with those from 35 NSCLC patients without oncogene-driven mutations received ICI (cohort B) or from 22 treatment-naïve NSCLC patients (cohort C). Data for each blood biomarker were summarized by mean and standard deviation and compared by Wilcoxon rank sum tests or Kruskal-Wallis tests with significance at 2-sided p value < 0.05. Co-culture of PBMCs and pleural effusion-derived tumor cells from individual patients with oncogene-driven NSCLC was used to determine the in vitro cytotoxicity of TKI and ICI.
Results
Except for low CD3% in cohort A, there were no significant differences in other 12 blood biomarkers among the 3 cohorts at baseline. TKI treatment in cohort A was associated with significant increase in CD3% and decrease in total and absolute neutrophils (p < 0.05). In cohort B, patients with good clinical response to ICI treatment (N = 18) had significant increases in absolute lymphocyte counts (ALCs), CD4 and/or CD8 cell counts. Conversely, those patients with poor clinical response to ICI (N = 17) had significant decreases in these cell counts. Of the 27 patients with pre- and post-treatment blood samples in cohort A, 11 had poor clinical response to TKIs and decreased lymphocyte counts. Of the remaining 16 patients who had good clinical response to TKI therapy, 10 (62.5%) patients had decreased, and 6 (37.5%) patients had increased lymphocyte counts. Multicolor immunophenoty** of PBMCs revealed ICI treatment activated additional immune cell types that need further validation. We confirmed that TKI treatment could either antagonize or enhance the effect of ICIs in the co-culture assay using patient’s tumor cells and PBMCs.
Conclusions
To the best of our knowledge, this is the first study showing that TKIs can have various effects on blood immune cells, which may affect their response to ICIs. Further validation of the blood biomarker and in vitro assay is warranted.
Similar content being viewed by others

Background
Immune checkpoint inhibitors (ICIs) have revolutionized the diagnosis and treatment for patients with locally advanced or metastatic non-small cell lung cancer (mNSCLC). However, ICIs have low or inferior clinical efficacy compared to chemotherapy in patients with epidermal growth factor receptor (EGFR)-mutant or anaplastic lymphoma kinase (ALK)-rearranged mNSCLC [1,2,3]. This low clinical efficacy of ICIs in EGFR-mutant or ALK-rearranged mNSCLC remains even when their tumors had high Programmed death-ligand 1 (PD-L1) immunohistochemistry (IHC) expression [4]. Furthermore, ICI treatment has been associated with increased incidence and severity of interstitial lung disease and immune-mediated adverse effects (including pneumonitis, colitis and hepatitis) when they are in sequential or concurrent use with small molecule tyrosine kinase inhibitors (TKIs) in patients with EGFR-mutant or ALK-rearranged mNSCLC [5]. Thus, mNSCLC patients with oncogene-driven mutations have been excluded in the first-line ICI trials except in the case of atezolizumab in IMpower 150 study [6]. The addition of atezolizumab to carboplatin, paclitaxel and bevacizumab (CPB) had superior clinical activity compared to CPB in a small cohort of patients with EGFR-mutant or ALK-rearranged cohorts after first-line TKI therapy. The key effect of angiogenesis inhibitor bevacizumab is postulated, which has synergism with each of the other 3 components. Recently, the updated report of PACIFIC [7] and retrospective analysis on durvalumab consolidation for patients with stage III NSCLC [8, 9] suggested that durvalumab might have limited clinical efficacy in the small subset of patients with EGFR- or HER2-mutant NSCLC.
ICIs are designed to activate exhausted tumor-reactive T cells, which are responsible for killing tumor cells. Current data suggest that the presence of high membranous PD-L1 IHC staining on tumor cells and the presence of intratumoral PD-1 expressing tumor infiltrating lymphocytes (TILs) in the tumor microenvironment (TME) are favourable prognostic factors and the best predictive factors of clinical response to ICIs [10]. The presence of a T-cell inflamed gene expression profile (GEP) in addition to PD-L1 IHC has improved the prediction of favourable clinical response to ICIs [10]. ICIs could increase the number of absolute lymphocyte counts (ALCs), restore the function in exhausted CD8+ T cells and induce phenotypically and functional changes of effector immune cells [11]. Several mechanisms have been postulated for the underlying mechanisms by which patients with oncogene-driven NSCLC do not derive clinical benefit from ICI [12]. These include low PD-L1 expression on tumor cells and TILs, low tumor mutation burden (TMB) and immune escape using other immune checkpoints in patients with EGFR-mutant NSCLC [13,14,27].
Data collection and statistical analysis
Data were summarized according to frequency and percentage for qualitative variables, and by mean ± standard deviation (SD) for quantitative variables unless noted otherwise. The 95% confidence interval for survivals was calculated using the exact binomial distribution. For each of the 13 blood cell types, the cell counts were summarized using mean and SD for pre- and post-treatment in cohort and response subgroups. Wilcoxon rank sum tests were used to compare two groups (or Kruskal-Wallis tests for three cohorts). Two-sided P < 0.05 was used to determine statistical significance. Due to the exploratory nature, adjustment for multiplicity was not performed for the types of blood cells [28]. Statistical analyses were carried out using SAS version 9.4 (SAS Institute, Cary, NC).
According to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 (http://ctep.info.nih.gov), lymphopenia (< 1000 cells/μl) was defined as grade 1: ALCs 800–999 cells/μL; grade 2: ALCs 500–799 cells/μL; grade 3: ALCs 200–499 cells/μL and grade 4: ALCs < 200 cells/μL. Best response to systemic therapies, defined as a complete or partial response (CR or PR), stable disease (SD) or progression disease (PD) achieved to cancer treatment, was assessed using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [29]. PFS was measured as the time from the first administration of a cancer therapy to progression defined by RECIST1.1, or death due to any cause. Patients alive without progression at the time of analysis were censored at the initiation of a new therapy or last follow-up. Good clinical response was defined in patients who achieved a CR, PR, PFS exceeding the reported median PFS for each targeted therapy. OS was measured as the time from the first administration of a cancer therapy to death due to any cause. Patients alive at the time of analysis were censored at the initiation of a new therapy or last follow-up. Survival data were estimated using the Kaplan–Meier method and compared using the log-rank test in each cohort and response subgroups. For the blood cell types significant in univariable survival analysis, multivariable survival analysis was further conducted using Cox proportional hazards models, by further adjusting for age, gender, race and histology in the model of each cell type.
Co-culture of patient’s tumor cells and PBMCs and growth inhibition by the MTS assay
H1975 and primary tumor cells isolated from the malignant pleural effusion of NSCLC patients with oncogene-driven mutations were seeded in 96-well plates at a density of 5 × 103 cells/well overnight. Human PBMCs from 20 ml of blood from the same patients were first purified by Ficoll-Paque and washed twice in 1x PBS. The acquired PBMCs were counted and added in a 2:1 ratio to the seeded tumor cells on the 96-well plates for co-culturing. After 8–12-h incubation, the cells were treated with various concentrations (0, 0.001, 0.01, 0.1, 1, 10 μM) of an TKI (osimertinib or alectinib) and/or an ICI (nivolumab (10 μg/mL), or atezolizumab (10 μg/mL) as indicated. The MTS assay was performed and analyzed as described previously [30, 31]. Specifically, after 72-h incubation, the MTS solution (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) was added and the cell viability was measured by the absorbance at 490 nm using a microplate reader (SpectraMax M3, Molecular Devices, USA). Untreated cells served as a control. Results were shown as the average cell viability ± SD [(ODtreat−ODblank)/(ODcontrol−ODblank) × 100%] of triplicate wells. Data were presented as the mean ± SD unless noted otherwise. All the experiments were performed in triplicate. Statistical analysis was performed using Graph Prism software (Version 8.21). Two-sided, P < 0.05 was considered statistically significant.
Results
Patients’ characteristics and baseline blood cell counts
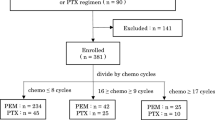
A total of 91 NSCLC patients were included in this study as illustrated in the flow chart (Fig. 1), 34 had blood samples after TKI treatment and 27 patients also had pre-treatment blood samples (cohort A), 35 had blood samples before and after ICI treatment (cohort B), and 22 had blood samples collected at diagnosis only (cohort C). Table 1 summarizes the clinicopathological and molecular characteristics of all study patients. Consistent with known clinicopathological features of oncogene-driven NSCLC, cohort A had more women (65%), Asians (50%), and lung adenocarcinomas (97%) compared to cohorts B and C. Driver oncogene mutations included EGFR mutations (N = 24, 70.6%), ALK fusions (N = 3, 8.8%), HER2 alterations (N = 3, 8.8%), MET alterations (N = 3, 8.8%) and RET fusion (N = 1, 2.9%). Details of 34 patients with oncogene-driven NSCLC are summarized in Table 2. Of 13 blood cell types, there were no significant differences in the baseline cell counts among the 3 cohorts of NSCLC patients except CD3% was significantly lower in cohort A (Table 3). However, we did not observe any significant differences among the absolute cell counts of major lymphocyte subtypes. Figure 2 illustrates the cell counts of 4 major blood immune cell types: ALCs (1.3 ± 0.77, 1.1 ± 0.56, and 1.2 ± 0.81 × 103 cells/μL for cohort A, B, C, respectively, P = 0.45) (Fig. 2a), CD4 counts (513 ± 283, 492 ± 288, and 599 ± 403 cells/μL for cohort A, B, C, respectively, P = 0.46) (Fig. 2b), CD8 (318 ± 242, 306 ± 260, and 327 ± 189 cells/μL for cohort A, B, C, respectively, P = 0.94) (Fig. 2c), and CD4 plus CD8 counts (831 ± 452, 798 ± 432, and 925 ± 556 cells/μL for cohort A, B, C, respectively, P = 0.61) (Fig. 2d). We did not find any correlation between immune cell counts and PD-L1 IHC expression or TMB (non-synonymous, somatic mutations per megabase, Mut/Mb) expression on tissues (Table 2).

Flow chart for study patients. Abbreviations: NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; NOS, not other specified; CR, complete response; mPFS, median progression free survival; PD, progression disease; PR, partial response

Assessment of baseline blood counts in patients with NSCLC. Baseline ALCs (A), CD4 (B), CD8 (C) and CD4 plus CD8 (D) cell counts for different cohorts are illustrated. Bar represents mean and SD. P-values were tested by the Kruskal-Wallis test
Cancer treatment modulated blood immune cells in NSCLC patients
Table 4 and Table 5 summarize the changes of 13 blood cell types in PBMCs in patients with mNSCLC with (Cohort A) or without (Cohort B) driver oncogenes, respectively, who had paired pre- and post-treatment blood samples. In cohort A (N = 27), TKI treatment was associated with significant decreases in white blood cells (WBCs, − 3.5 ± 1.1 × 103 cells/μL, P = 0.015), absolute neutrophil counts (ANCs, − 3.6 ± 1.1 cells/μL, P = 0.014), and dNLR (− 2.4 ± 1.2, P = 0.027) but increased CD3% (6.4 ± 2.5%, P = 0.041) in poor responders versus good responders (Table 4). Figure 3 illustrates the major changes in ALCs, CD4, CD8, and CD4 plus CD8 cell counts in good responders (left panel) and poor response (right panel), respectively. In cohort A, the patients with oncogene-driven NSCLC received targeted therapy with specific TKI. Flow cytometry analysis showed that TKI treatment had various effects on the blood immune cells (Fig. 3a, c, e, g). Of 16 patients with good clinical response to TKI therapy (left panel), 10 (62.5%) patients had decreased ALCs and 6 (37.5%) patients had increased ALCs (Fig. 3a, left). Similar changes were observed in post-treatment CD4 counts (Fig. 3c, left), CD8 counts (Fig. 3e, left), and CD4 plus CD8 counts (Fig. 3g, left), respectively, in patients with good clinical responses to TKI treatment. In contrast, all 11 patients with poor clinical response to TKIs had significantly decrease in ALCs (from 1200 ± 900 to 900 ± 400 × 103 cells/μL, P = 0.01) (Fig. 3a, right), the CD4 cell counts (from 472 ± 245 to 345 ± 180 cells/μL, P = 0.003) (Fig. 3c, right), the CD8 cell counts (from 240 ± 98 to 184 ± 89 cells/μL, P = 0.001) (Fig. 3e, right), and the CD4 plus CD8 cell counts (from 711 ± 301 to 529 ± 227 cells/μL, P = 0.002) (Fig. 3g, right), respectively. These results suggested that small molecule TKIs may modulate blood immune cell count in patients with oncogene-driven NSCLC.

Comparison of changes in ALCs, CD4, CD8 and CD4 plus CD8 cell counts pre- and after-treatment. ALCs, CD4, CD8, and CD4 plus CD8 counts by flow cytometry in good responders and poor responders in NSCLC patients receiving TKI treatment (Cohort A) (A, C, E, G) or receiving ICI (Cohort B) (B, D, F, H) are shown. Groups were compared by the Wilcoxon signed rank test. *P < 0.05 for statistical significance. Abbreviations: ALCs, absolute lymphocyte cells; ICI, immune checkpoint inhibitors
In cohort B (N = 35), the patients without oncogene-driven NSCLC received a PD-(L)1 ICI (i.e., pembrolizumab, nivolumab, or atezolizumab), either alone (N = 14) or in combination with chemotherapy (N = 21) (Table 5). Compared to pre-treatment, ICI treatment was associated with significant increases in post-treatment ALCs (from 1100 ± 600 to 1300 ± 800 × 103 cells/μL, P < 0.001) (Fig. 3b, left), CD4 counts (from 503 ± 284 to 593 ± 297 cells/μL, P = 0.004) (Fig. 3d, left), CD8 counts (from 280 ± 323 to 400 ± 476 cells/μL, P = 0.006) (Fig. 3f, left), and CD4 plus CD8 cell counts (from 782 ± 483 to 994 ± 627 cells/μL, P < 0.001) (Fig. 3h, left), respectively, in good responders (N = 18). In contrast, ICI treatment was associated with significant decreases in post-treatment in ALCs (from 1100 ± 500 to 800 ± 300 × 103 cells/μL, P < 0.001) (Fig. 3b, right), CD4 counts (from 481 ± 300 to 311 ± 204 cells/μL, P = 0.004) (Fig. 3d, right), CD8 counts (from 333 ± 178 to 215 ± 106 cells/μL, P = 0.006) (Fig. 3f, right), and CD4 plus CD8 counts (from 814 ± 384 to 526 ± 251 cells/μL, P < 0.001) (Fig. 3h, right), respectively, in poor responders to ICI (n = 17). There results were comparable to patients with oncogene-driven NSCLC who had poor clinical response to TKI (Fig. 3, cohort A, right).
Post-treatment lymphopenia was associated with poor clinical benefit in NSCLC patients
With a median follow-up of 24.5 months, there were no significant differences in median PFS (10.6 vs. 5.5 months, P = 0.20) and OS (25.3 vs. 25.8 months, P = 0.94) in cohort A and cohort B (Fig. 4). The correlation of the post-treatment ALCs, CD4 plus CD8 counts, PD-L1 IHC, and TMB were determined with the clinical outcomes in each cohort of NSCLC patients. Currently, there is no established cutoff for ALCs as a biomarker [19,20,21,22]. Using the receiver operating characteristic (ROC) curve and Youden index analysis, we identified 800 and 500 cells/μl as the optimal cut-off values for ALCs and CD4+ plus CD8+ cell counts, respectively (Fig. 4c). Figure 5 illustrates the median PFS and OS according to ALCs, CD4 plus CD8 counts, PD-L1 IHC, and TMB in cohort A patients (N = 27). Compared to those patients with post-TKI treatment ALCs < 800 cells/μL, patients with post-TKI treatment ALCs ≥800 cells/μL had longer median PFS (16.0 vs. 4.4 months; HR 5.08, 95% CI 1.62–15.92, P = 0.0023) (Fig. 5A1) and longer median OS (26.2 vs. 10.7 months; HR 10.15, 95% CI 2.46–41.76, P < 0.0001) (Fig. 5B1), respectively. Similarly, patients with post-TKI treatment CD4 plus CD8 counts ≥500 cells/μL had statistically better PFS and OS compared to those patients with CD4 plus CD8 counts < 500 cells/μL (Fig. 5A2 and B2). In contrast, PD-L1 IHC and TMB expression in patients with oncogene-driven NSCLC did not correlate with the PFS (Fig.5A3 and A4) and OS (Fig. 5B3 and 5B4). Multivariate analysis using the Cox proportional hazards regression model showed ALCs remained significant for PFS (P = 0.024, HR 4.25, 95% CI 1.2–14.9) and OS (P = 0.022, HR 7.59, 95% CI 1.33–43.16), while CD4 plus CD8 counts was only significant for PFS (P = 0.006, HR 8.89, 95% CI 1.89–41.86) in cohort A (Tables 6 and 7).

Kaplan-Meier PFS and OS estimates by lymphocyte counts according to NSCLC cohorts. Median PFS (A) and OS (B) of NSCLC patients are illustrated according to cohort A and B, respectively. Groups were compared using the log-rank test. Receiver operating characteristic (ROC) curves analysis for ALCs (C) and CD4 plus CD8 (D) in enrolled NSCLC patients (N = 62) are shown. Tick marks indicate censored data. Groups were compared using the Z test. P < 0.05 for statistical significance

Kaplan-Meier PFS and OS estimates according to ALCs, CD4 plus CD8 counts, PD-L1 IHC and TMB in cohort A. Post-treatment low ALCs (< 800 cells/μL, red) were associated with shorter PFS and OS compared to high ALCs (≥800 cells/μL, blue) in patients with oncogene-driven NSCLC (cohort A) (A and B), respectively. Post-treatment low CD4 plus CD8 cell counts (< 500 cells/μL, red) were associated with shorter PFS and OS compared to high CD4 plus CD8 cell counts (≥500 cells/μL, blue) (cohort A) (A and B), respectively. PD-L1 IHC expression (C) and TMB status (D) did not affect the PFS and OS in NSCLC patients with oncogene driven NSCLC. Tick marks indicate censored data. Groups were compared using the log-rank test. P < 0.05 for statistical significance
Figure 6 illustrates the median PFS and OS according to ALCs, CD4 plus CD8 counts, PD-L1 IHC, and TMB in cohort B patients (N = 35). Patients without oncogene-driven NSCLC whose post-ICI ALCs ≥800 cells/μL had significantly longer median PFS (6.6 vs. 4.3 months, HR 2.63, 95% CI 1.16–5.99, P = 0.016) (Fig. 6A1) but not median OS benefit (27.8 vs. 21.3 months, HR 2.38, 95% CI 0.89–6.35, P = 0.076) (Fig. 6B1). Patients with CD4 plus CD8 counts ≥500 cells/μL had significantly longer median PFS (6.5 vs 3.5 months, HR 2.7, 95% CI: 1.21–6.04, P = 0.012) (Fig. 6A2) and longer median OS (27.8 vs. 11.2 months, HR 3.03, 95% CI 1.12–8.2, P = 0.026) (Fig. 6B2). Patients with PD-L1 IHC ≥50% had significantly longer median PFS (7.8 vs. 5.2 months, HR 2.36, 95% CI: 1.04–5.37, P = 0.034) (Fig. 6A3) and longer median OS (37.9 vs. 18.5 months, HR 3.18, 95% CI 0.97–10.45, P = 0.047) (Fig. 6B3). Patients with TMB expression ≥10 mut/mb had significantly longer median PFS (7.9 vs. 4.7 months, HR 3.16, 95% CI: 1.27–7.86, P = 0.009) (Fig. 6A4). In contrast, TMB expression did not correlate with OS (Fig. 6B4) in patients without oncogene-driven NSCLC. Multivariate analysis using the Cox proportional hazards regression model revealed that ALCs, CD4 plus CD8 counts, PD-L1 IHC and TMB are an independent significant predictor for PFS (P = 0.038, P = 0.021, P = 0.003 and P = 0.004, respectively), and CD4 plus CD8 counts remained a significant predictor for OS (P = 0.008, HR 5.96, 95% CI 1.60–22.2), for NSCLC patients without driver oncogenes receiving ICI treatment (Tables 6 and 7).

Kaplan-Meier PFS and OS estimates according to ALCs, CD4 plus CD8 counts, PD-L1 IHC and TMB in cohort B. In patients without oncogene-driven NSCLC (cohort B), post-treatment low ALCs (< 800 cells/μL, red) were associated with statistically significant shorter PFS (A) but not OS (B) compared to high ALCs (≥800 cells/μL, blue). Post-treatment low CD4 plus CD8 cell counts (< 500 cells/μL, red) were associated with shorter PFS and OS compared to high CD4 plus CD8 cell counts (≥500 cells/μL, blue) (cohort B) (A and B), respectively. Patients with NSCLC expressing PD-L1 IHC < 50% had shorter PFS and shorter OS compared to those patients with NSCLC expressing PD-L1 IHC ≥50% (C). Patients with NSCLC expressing TMB ≥10 Mut/Mb had shorter PFS but not OS compared to those patients with NSCLC expressing TMB < 10 Mut/Mb (D). Tick marks indicate censored data. Groups were compared using the log-rank test. P < 0.05 for statistical significance
Immunophenotypic changes of PBMCs in a patient who received ICI combination
Figure 7a summarizes the clinical course of a patient with EGFR exon 19 deletion who received ICIs at diagnosis when tumor genomic profiling test did not identify the EGFR mutation. Patient subsequently received platinum-based chemotherapy for 2 cycles and has been having clinical PR to EGFR TKI osimertinib. The changes in ALCs, CD4, CD8, and CD4 plus CD8 cell counts during the treatment course were shown in Fig. 7c. Although CD4+ and CD8+ T cells account for the majority of ALCs, ALCs include a few rare yet important lymphocyte subtypes such as natural killer (NK) cells and monocytes. In addition, changes in these lymphocytes might affect other immune cell types such as B cells in the PBMCs. Further, the immunophenotypic analysis of PBMCs collected before and after ICI treatment were performed for extended immune cell types, including B cells, NK cells, monocytes, using a multicolor flowcytometry analysis. As showed in heatmap (Figs. 7b) and viSNE land (Figs. 7d), the ICI treatment increased lymphocytes, CD3+, CD4+, CD4 TEMRA, CD8+, CD8 central memory, CD8 naïve cells and NK cells and decreased all other types of immune subtype cells including PD1 + CD4 and PD1 + CD8 cells.

Immunophenotypic changes of PBMCs in a patient who received ICI combination. A summarizes the key events in the clinical course of a patient with EGFR exon 19 deletion. A 72-year-old gentleman, never smoker was diagnosed with cT4N2M1c NSCLC adenocarcinoma with large tumor burden in April 2020 when he presented with worsening non-productive cough and shortness of breath for about 6 months. The tumor was stained negative for PD-L1 IHC stain (DAKO, clone 22C3). Tumor genomic profiling did not identify any driver oncogenes. The patient received PD-1 and CTLA-4 inhibitors for 3 cycles and was found to have tumor progression. The patient subsequently received carboplatin and pemetrexed for two cycles without significant clinical improvement. Repeat tumor genomic profiling test identified an EGFR E19Del. The patient had been in clinical PR to EGFR TKI osimertinib for over 10 months at the time of this report. The changes in ALCs, CD4, CD8, and CD4 plus CD8 cell counts during the treatment course (B). Heat map** (C) and viSNE land (D) illustrate the immunophenotypic changes of PBMCs before and after ICI treatment
Patients with oncogene-driven NSCLCs have various responses to ICIs
We developed an in vitro cytotoxic assay using patient’s malignant pleural effusion and PBMCs to determine the effect of small molecule TKIs and ICIs on patient tumor cells and PBMCs (Fig. 8). As shown in Fig. 8a, osimertinib, nivolumab and atezolizumab inhibited the growth of H1975 cells by 28 ± 13%, 77 ± 28% and 66 ± 21%, respectively. In patient-derived, EGFR-mutant lung adenocarcinoma cells that were resistant to osimertinib (growth inhibition of 81 ± 15%), nivolumab and atezolizumab significantly inhibited the growth by (65 ± 18%) and (57 ± 13%), respectively, P < 0.05) (Fig. 8b). The addition of atezolizumab to osimertinib significantly inhibited the growth of both H1975 cells (11 ± 6%, P < 0.001) (Fig. 8a) and patient derived, EGFR-mutant NSCLC cells (8 ± 1%), (P < 0.001) (Fig. 8b). Osimertinb and atezolizumab had stronger cytotoxic synergism compared to osimertinib and nivolumab in both osimertinib-sensitive, EGFR-mutant (11 ± 6% vs 25 ± 6%, P < 0.05) (Fig. 8a) and osimertinib-resistant, EGFR-mutant lung adenocarcinomas (8 ± 1% vs 28 ± 3%, P < 0.05) (Fig. 8b).

Growth inhibition of malignant tumor cells and PBMCs from the same patients with oncogene-driven NSCLC by TKIs and ICIs. Tumor cells isolated from malignant pleural effusion and PBMCs isolated from the peripheral blood of individual patients were cocultured for 12 h before treated with specific TKI and/or ICIs. H1975 cells (A) and osimertinib-resistant NSCLC cells (B) were cultured with patient’s PBMCs, and treated with vehicle, osimertinib (0.1 μM), nivolumab (10 μg/ml), atezolizumab (10 μg/ml) or combination as indicated for 72 h. Growth inhibition was measured using the MTS assay using vehicle as 100% control. Alectinib-sensitive ALK-fusion NSCLC cells (C) and alectinib-resistant RET-fusion NSCLC cells (D) were cocultured with PBMCs from corresponding patients and treated with vehicle, alectinib (0.1 μM), nivolumab (10 μg/ml), atezolizumab (10 μg/ml) or combination as indicated for 72 h. Growth inhibition was measured using the MTS assay with vehicle as 100% control. All data are shown as mean of triplicate samples. Error bars indicate standard deviation (SD). Groups were compared by the Wilcoxon signed rank test. P < 0.05 was considered statistically significant. Abbreviations: MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
ALK inhibitor alectinib, nivolumab and atezolizumab inhibited the growth of alectinib-sensitive, patient-derived EML4-ALK fusion NSCLC cells by 29 ± 14%, 53 ± 15%, 65 ± 13%, respectively. Although the addition of nivolumab to alectinib did not significantly increase the growth of tumor cells compared to alectinib or nivolumab alone (21 ± 6%, 29 ± 14%, vs 53 ± 15%, respectively), and the addition of atezolizumab to alectinib had antagonistic effect on tumor cell growth compared to alectinib alone (46 ± 4% vs 29 ± 14%, respectively) (Fig. 8c). In alectinib-resistant RET fusion NSCLC cells from another patient, nivolumab or atezolizumab, either alone or in combination with alectinib, did not significant inhibit the growth of tumor cells (102 ± 11%, 75 ± 4%, 74 ± 6%, 79 ± 3%, 75 ± 9% respectively) (Fig. 8d). These data support that patients with oncogene-driven NSCLCs could have various responses to ICIs either alone or in combination with TKIs. However, we could not verify the clinical response of ICI and ICI-TKI combination in patients as these were contraindicated clinically.
Discussion
Our study has several clinical implications. First, lymphocyte counts and immunophenoty** of T-cell and B-cell have been used to assess the immune status and are prognostic biomarkers. Low levels of lymphocytes in the blood could indicate an increased risk for death [32]. The American Society of Hematology (ASH)-American Society for Transplantation and Cellular Therapy (ASTCT) defined patients with neutropenia ANC < 500 cells/μL and lymphopenia ALCs< 200 cells/μL as immunodeficiencies and recommended against COVID-19 vaccination [33]. We found that all study patients had ALCs > 200/μL during almost the entire disease course (except 3 patients at imminent dying stage), which is safe for receiving COVID-19 vaccination [34]. It is well known that many cancer treatments for hematological malignancies are immunosuppressive, which include cytotoxic chemotherapy, monoclonal antibodies against CD20, CD38 and CD52, calcineurin inhibitors, Mammalian Target of Rapamycin (mTOR), bortezomib and Bruton’s tyrosine kinase (BTK) inhibitors [35, 36]. A recent study showed that severe lymphopenia (ALCs < 500 cells/μL) before starting the consolidation durvalumab in patients with unresected locally advanced NSCLC after completion of definitive chemoradiation was associated with worse PFS compared to those patients without severe lymphopenia [37]. Although immune modulation is well documented in targeted therapy with monoclonal antibody for solid tumors [38], there is no prior study to determine the effect of TKIs on blood immune cells in patients with oncogene-driven NSCLC. In large clinical trials of TKIs for patients with advanced oncogene-driven NSCLC, the incidence and severity of leukopenia, neutropenia, and lymphopenia are generally less than 5% (Table 6) [39,40,41,42,43,44,45,46,47,48,49]. We observed that at baseline the patients with oncogene-driven NSCLC in cohort A had lower CD3% compared to patients without oncogene-driven NSCLC in cohort B and control group in cohort C. After TKI treatment, and these patients in cohort A had significantly increases in CD3% and decreased WBC, ANC and dLNR (Table 4). Consistent with previous reports, we also found that cancer progression [50] cytoreduction by chemotherapy and/or radiation [51,52,53,54], infection [32], and steroids use [55] were common reasons associated with decreased lymphocyte counts in patients with mNSCLC. This could reverse promptly with the clinical improvement or stop** steroids. While the changes of immune cell counts were associated with good or poor clinical responses to ICIs, targeted therapy with TKIs could modulate immune cell counts that mimic good or poor clinical responses to ICIs. The impact of our observation on patient’s response to ICIs is unknown and needs further exploration.
Second, small molecule TKIs targeting a growing number of gain-of-function molecular targets, such as EGFR, BRAF V600E, MET exon 14 skip** mutations, and ALK-, ROS1-, and neurotrophic receptor tyrosine kinase (NTRK)- or RET-gene fusions have been shown to improve PFS and OS with favorable toxicity profiles compared to platinum-containing combination chemotherapy as first-line systemic therapy in ~ 25% of NSCLC patients (Table 8). It is important to understand the role of TKIs on the immune system and treatment with ICIs. Compared to molecular biomarkers, immune biomarkers are complex with many different components that are subjected to change during natural tumor progression and treatment. A coordinated response by both humoral immunity and cell-mediated immunity is important to the response to ICIs [64, 65]. Using multiplex flow cytometry, we performed the phenotypic analysis of various immune cells in the patient PBMCs. These immune cells include T cell subsets (such as effector, activated, memory, exhausted, and regulatory), B cells, and NK/NKT cells. CD8+ T cells are the primary effector cells against tumors. The presence of activated, circulating, tumor-derived, PD1 + CD8+ T cells in patient PBMCs have been associated with clinical response to ICI therapy. The tumor-antigen specificities and TCR repertoires of the circulating and tumor-infiltrating CD8 + PD-1+ cells appeared similar. We observed that CD4 plus CD8 counts were an independent biomarker for PFS of NSCLC patients in both cohort A and B, and OS of NSCLC patients in cohort B. ALCs were an independent biomarker for OS in patients with oncogene-driven NSCLC. In cohort B, post-treatment CD4 plus CD8 cell counts performed better than the known immune biomarkers (PD-L1 IHC and TMB) in predicting response to ICIs. Further characterization of the effect of TKIs on the expression and function of immune cells in oncogene-driven NSCLC are warranted.
Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC, implicating an optional immune targeted therapy for NSCLC patients with EGFR mutation. In genetically engineered mouse models (GEMMs), EGFR-driven tumors express higher levels of PD-L1 with a more immunosuppressive tumor microenvironment (increased FoxP3+ T-cells, decreased CD8+/CD4+ ratio). The addition of an EGFR-TKI in these EGFR-mutant GEMM models modulated PD-L1 expression and reversed EGFR-pathway mediated immunosuppression. However, ICIs either alone or in combination with small molecule TKIs have low or inferior effect in EGFR-mutant or ALK-rearranged NSCLC. Therefore, GEMMs are not good models to study the effect of ICIs on human NSCLC tumors. As PD-L1 expression is lower in the majority of oncogene-driven NSCLC compared to non-oncogene-driven NSCLC, this may explain why the results in GEMMs did not correlate with the clinical observation. We also did not observe significant predictive or prognostic association between lymphocyte counts and correlation of PD-L1 or TMB. Using multiplex flow cytometry, we evaluated the immunophenotypic changes of PBMCs in a patient with EGFR E19 deletion who received 3 cycles of ICIs. We found that the ICI treatment increased lymphocytes, CD4+, CD4 terminally differentiated effector memory (TEMRA; CD45RA+ CCR7-), and CD8 naïve cells, and decreased all other types of immune subtype cells, including PD1 + CD4 and PD1 + CD8 cells. Of note, NSCLC patients with oncogene-driven mutations, such as EGFR, have been associated with hyperprogression to ICIs [66]. The impact of our observation and the association of blood circulating immune cells with TILs in TME in patients with oncogene-driven NSCLC after ICI treatment is unknown and deserves further exploration.
Malignant biofluids such as pleural effusion is a unique source for liquid biopsy that is currently underused for molecular diagnosis and tumor biology study. Pleural effusion affects at least 40% of patients with lung cancer. Up to 90% of these patients have confirmed malignant pleural effusion (MPE) and require palliative thoracentesis for symptomatic relief [67]. We explored the effect of TKIs on patient’s tumor cells from malignant pleural effusion and PBMCs using an in vitro co-culture model. We observed significant variations in individual’s response to different ICIs that were not clearly associated with tumor PD-L1 expression or sensitivity to targeted therapy with TKIs. To the best of our knowledge, this is the first study to determine the effect of small molecule targeted therapy on lymphocyte cells using NSCLC patient’s blood samples and malignant tumor cells. Ongoing study is determining the clinical utility of this in vitro assay in predicting clinical response to ICI therapy in patients with oncogene-driven NSCLC.
There are several limitations to this study, including its small sample size, it being a retrospective study, and no adjustment for multiplicity due to the exploratory nature. The potential selection bias and the imbalance of the baseline characteristics and treatment history in patients may have contributed to the treatment outcomes. Furthermore, dysregulation of various immunoregulatory cells and cytokines in the TME may be responsible for tumor response [68, 69]. We did not perform the functional analysis of immune subtypes in patient PBMCs collected before and after TKI or ICI treatment. Further study is needed to confirm our findings and understand the effect of TKI modulation on “cold” or “hot” tumor microenvironment.
Conclusions
Molecularly targeted therapy by small molecule TKIs have various effects on modulating the blood immune cell count in patients with oncogene-driven NSCLC. There are unmet needs to understand the underpinning mechanisms and develop predictive biomarkers and assays to select the appropriate patients for ICI therapy.
Availability of data and materials
All data supporting the conclusions of this research article are included within the manuscript.
Abbreviations
- ALCs:
-
Absolute lymphocyte counts
- ALK:
-
Anaplastic lymphoma kinase
- ANC:
-
Absolute neutrophil counts
- ASH:
-
American Society of Hematology
- ASTCT:
-
American Society for Transplantation and Cellular Theory
- BTK:
-
Bruton tyrosine kinase
- CPB:
-
Carboplatin, paclitaxel, bevacizumab
- CR:
-
Complete response
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- dNLR:
-
derives neutrophil-to-lymphocyte ratio
- EGFR:
-
Epidermal growth factor receptor
- GEP:
-
Gene expression profile
- EDTA:
-
Ethylenediamine tetraacetic acid
- GEMMs:
-
Genetically engineered mouse models
- HER2:
-
Human epidermal growth factor receptor 2
- ICIs:
-
Immune checkpoint inhibitors
- IHC:
-
Immunohistochemistry
- IRB:
-
Institutional Review Board
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- mTOR:
-
mammalian target of rapamycin
- MPE:
-
Malignant pleural effusion
- MTS:
-
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
- mNSCLC:
-
metastatic non-small cell lung cancer
- N:
-
Number
- NSCLC:
-
Non-small cell lung cancer
- NGS:
-
Next generation sequencing
- NCCN:
-
National Comprehensive Cancer Network
- NCI:
-
National Cancer
- NK:
-
Natural killer
- NOS:
-
Not otherwise specified
- OS:
-
Overall survival
- PBMCs:
-
Peripheral blood mononuclear cells
- PBS:
-
Phosphate-buffered saline
- PD:
-
Progression disease
- PD-L1:
-
Programmed death-ligand 1
- PFS:
-
Progression free survival
- PR:
-
Partial response
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- ROC:
-
Receiver operating characteristic
- SD:
-
Stable disease
- SD:
-
Standard deviation
- TCR:
-
T-cell receptor
- TKIs:
-
Tyrosine kinase inhibitors
- TILs:
-
Tumor infiltrating lymphocytes
- TMB:
-
Tumor mutation burden
- TME:
-
Tumor micro environment
- WBC:
-
White blood cell
References
Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung Cancer: a retrospective analysis. Clin Cancer Res. 2016;22(18):4585–93. https://doi.org/10.1158/1078-0432.CCR-15-3101.
Oxnard GR, Yang JC, Yu H, Kim SW, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31(4):507–16. https://doi.org/10.1016/j.annonc.2020.01.013.
Gavralidis A, Gainor JF. Immunotherapy in EGFR-mutant and ALK-positive lung Cancer: implications for oncogene-driven lung Cancer. Cancer J. 2020;26(6):517–24. https://doi.org/10.1097/PPO.0000000000000491.
Lisberg A, Cummings A, Goldman JW, Bornazyan K, Reese N, Wang T, et al. A phase II study of Pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naive patients with advanced NSCLC. J Thorac Oncol. 2018;13(8):1138–45. https://doi.org/10.1016/j.jtho.2018.03.035.
Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and Meta-analysis. JAMA Oncol. 2018;4(2):210–6. https://doi.org/10.1001/jamaoncol.2017.4427.
Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7(5):387–401. https://doi.org/10.1016/S2213-2600(19)30084-0.
Faivre-Finn C, Vicente D, Kurata T, Planchard D, Paz-Ares L, Vansteenkiste JF, et al. Four-year survival with Durvalumab after Chemoradiotherapy in stage III NSCLC-an update from the PACIFIC trial. J Thorac Oncol. 2021;16(5):860–7. https://doi.org/10.1016/j.jtho.2020.12.015.
Aredo JV, Mambetsariev I, Hellyer JA, Amini A, Neal JW, Padda SK, et al. Durvalumab for stage III EGFR-mutated NSCLC after definitive Chemoradiotherapy. J Thorac Oncol. 2021;16(6):1030–41. https://doi.org/10.1016/j.jtho.2021.01.1628.
Hellyer JA, Aredo JV, Das M, Ramchandran K, Padda SK, Neal JW, et al. Role of consolidation Durvalumab in patients with EGFR- and HER2-mutant Unresectable stage III NSCLC. J Thorac Oncol. 2021;16(5):868–72. https://doi.org/10.1016/j.jtho.2020.12.020.
Chen JA, Ma W, Yuan J, Li T. Translational biomarkers and rationale strategies to overcome resistance to immune checkpoint inhibitors in solid tumors. Cancer Treat Res. 2020;180:251–79. https://doi.org/10.1007/978-3-030-38862-1_9.
Nakamura Y. Biomarkers for immune checkpoint inhibitor-mediated tumor response and adverse events. Front Med. 2019;6:119. https://doi.org/10.3389/fmed.2019.00119.
Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung Cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–41. https://doi.org/10.1200/JCO.2017.75.3384.
Soo RA, Kim HR, Asuncion BR, Fazreen Z, Omar MFM, Herrera MC, et al. Significance of immune checkpoint proteins in EGFR-mutant non-small cell lung cancer. Lung Cancer. 2017;105:17–22. https://doi.org/10.1016/j.lungcan.2017.01.008.
Lin A, Wei T, Meng H, Luo P, Zhang J. Role of the dynamic tumor microenvironment in controversies regarding immune checkpoint inhibitors for the treatment of non-small cell lung cancer (NSCLC) with EGFR mutations. Mol Cancer. 2019;18(1):139. https://doi.org/10.1186/s12943-019-1062-7.
Dong ZY, Zhang JT, Liu SY, Su J, Zhang C, **e Z, et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology. 2017;6(11):e1356145. https://doi.org/10.1080/2162402X.2017.1356145.
Biton J, Mansuet-Lupo A, Pecuchet N, Alifano M, Ouakrim H, Arrondeau J, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin Cancer Res. 2018;24(22):5710–23. https://doi.org/10.1158/1078-0432.CCR-18-0163.
Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014;25(10):1935–40. https://doi.org/10.1093/annonc/mdu242.
**ao W, Ma W, Wei S, Li Q, Liu R, Carney RP, et al. High-affinity peptide ligand LXY30 for targeting alpha3beta1 integrin in non-small cell lung cancer. J Hematol Oncol. 2019;12(1):56. https://doi.org/10.1186/s13045-019-0740-7.
Ho WJ, Yarchoan M, Hopkins A, Mehra R, Grossman S, Kang H. Association between pretreatment lymphocyte count and response to PD1 inhibitors in head and neck squamous cell carcinomas. J Immunother Cancer. 2018;6(1):84. https://doi.org/10.1186/s40425-018-0395-x.
Karantanos T, Karanika S, Seth B, Gignac G. The absolute lymphocyte count can predict the overall survival of patients with non-small cell lung cancer on nivolumab: a clinical study. Clin Transl Oncol. 2019;21(2):206–12. https://doi.org/10.1007/s12094-018-1908-2.
Pan M, Alavi M, Herrinton LJ. Association of Inflammatory Markers with disease progression in patients with metastatic melanoma treated with immune checkpoint inhibitors. Perm J. 2018;22:17–149. https://doi.org/10.7812/TPP/17-149.
Huemer F, Lang D, Westphal T, Gampenrieder SP, Hutarew G, Weiss L, et al. Baseline absolute Lymphocyte count and ECOG performance score are associated with survival in advanced non-small Cell Lung Cancer undergoing PD-1/PD-L1 Blockade. J Clin Med. 2019;8(7):1014.
Park JC, Durbeck J, Clark JR. Predictive value of peripheral lymphocyte counts for immune checkpoint inhibitor efficacy in advanced head and neck squamous cell carcinoma. Mol Clin Oncol. 2020;13(6):87. https://doi.org/10.3892/mco.2020.2157.
Naidus E, Bouquet J, Oh DY, Looney TJ, Yang H, Fong L, et al. Early changes in the circulating T cells are associated with clinical outcomes after PD-L1 blockade by durvalumab in advanced NSCLC patients. Cancer Immunol Immunother. 2021;70(7):2095–102. https://doi.org/10.1007/s00262-020-02833-z.
O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67. https://doi.org/10.1038/s41571-018-0142-8.
Gavrilov S, Zhudenkov K, Helmlinger G, Dunyak J, Peskov K, Aksenov S. Longitudinal tumor size and neutrophil-to-lymphocyte ratio are prognostic biomarkers for overall survival in patients with advanced non-small cell lung Cancer treated with Durvalumab. CPT Pharmacometrics Syst Pharmacol. 2021;10(1):67–74. https://doi.org/10.1002/psp4.12578.
Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom. 2010;10:10–7.
Li G, Taljaard M, Van den Heuvel ER, Levine MA, Cook DJ, Wells GA, et al. An introduction to multiplicity issues in clinical trials: the what, why, when and how. Int J Epidemiol. 2017;46(2):746–55. https://doi.org/10.1093/ije/dyw320.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47. https://doi.org/10.1016/j.ejca.2008.10.026.
Ma W, Gong J, Shan J, Lewis D, **ao W, Moore EH, et al. Safety and efficacy of Osimertinib in the treatment of a patient with metastatic lung Cancer and concurrent somatic EGFR L858R and germline EGFR T790M mutations. JCO Precision Oncol. 2018;2:1–7.
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10(6):910–23. https://doi.org/10.1097/JTO.0000000000000500.
Warny M, Helby J, Nordestgaard BG, Birgens H, Bojesen SE. Lymphopenia and risk of infection and infection-related death in 98,344 individuals from a prospective Danish population-based study. PLoS Med. 2018;15(11):e1002685. https://doi.org/10.1371/journal.pmed.1002685.
General Principles of COVID-19 Vaccines for Immunocompromised. (Version 3.0; last updated April 5, 2021) Patients.https://www.hematology.org/covid-19/ash-astct-covid-19-and-vaccines. Accessed 28 Apr 2021.
NCCN: Cancer and COVID-19: Recommendations of the NCCN COVID-19 Vaccination Advisory Committee Vaccination.https://www.nccn.org/covid-19/pdf/COVID-19_Vaccination_Guidance_V2.0.pdf. accessed 3/10/2021.
Mikulska M, Lanini S, Gudiol C, Drgona L, Ippolito G, Fernandez-Ruiz M, et al. ESCMID study Group for Infections in compromised hosts (ESGICH) consensus document on the safety of targeted and biological therapies: an infectious diseases perspective (agents targeting lymphoid cells surface antigens [I]: CD19, CD20 and CD52). Clin Microbiol Infect. 2018;24(Suppl 2):S71–82. https://doi.org/10.1016/j.cmi.2018.02.003.
Allegrezza MJ, Conejo-Garcia JR. Targeted therapy and immunosuppression in the tumor microenvironment. Trends Cancer. 2017;3(1):19–27. https://doi.org/10.1016/j.trecan.2016.11.009.
Friedes C, Chakrabarti T, Olson S, Prichett L, Brahmer JR, Forde PM, et al. Association of severe lymphopenia and disease progression in unresectable locally advanced non-small cell lung cancer treated with definitive chemoradiation and immunotherapy. Lung Cancer. 2021;154:36–43. https://doi.org/10.1016/j.lungcan.2021.01.022.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. https://doi.org/10.1038/s41577-020-0306-5.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32. https://doi.org/10.1056/NEJMoa050753.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703. https://doi.org/10.1056/NEJMoa1006448.
Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–34. https://doi.org/10.1200/JCO.2012.44.2806.
Mok TS, Wu YL, Papadimitrakopoulou VA. Osimertinib in EGFR T790M-positive lung Cancer. N Engl J Med. 2017;376(20):1993–4. https://doi.org/10.1056/NEJMc1703339.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung Cancer. N Engl J Med. 2018;378(2):113–25. https://doi.org/10.1056/NEJMoa1713137.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with Osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. https://doi.org/10.1056/NEJMoa1913662.
Wu YL, Herbst RS, Mann H, Rukazenkov Y, Marotti M, Tsuboi M. ADAURA: phase III, double-blind, randomized study of Osimertinib versus placebo in EGFR mutation-positive early-stage NSCLC after complete surgical resection. Clin Lung Cancer. 2018;19(4):e533–6. https://doi.org/10.1016/j.cllc.2018.04.004.
Wu YL, Tsuboi M, He J, John T, Grohe C, Majem M, et al. Osimertinib in resected EGFR-mutated non-small-cell lung Cancer. N Engl J Med. 2020;383(18):1711–23. https://doi.org/10.1056/NEJMoa2027071.
Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus Crizotinib in untreated ALK-positive non-small-cell lung Cancer. N Engl J Med. 2017;377(9):829–38. https://doi.org/10.1056/NEJMoa1704795.
Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung Cancer. N Engl J Med. 2020;383(10):944–57. https://doi.org/10.1056/NEJMoa2002787.
Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of Selpercatinib in RET fusion-positive non-small-cell lung Cancer. N Engl J Med. 2020;383(9):813–24. https://doi.org/10.1056/NEJMoa2005653.
Ray-Coquard I, Cropet C, Van Glabbeke M, Sebban C, Le Cesne A, Judson I, et al. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 2009;69(13):5383–91. https://doi.org/10.1158/0008-5472.CAN-08-3845.
Tang C, Liao Z, Gomez D, Levy L, Zhuang Y, Gebremichael RA, et al. Lymphopenia association with gross tumor volume and lung V5 and its effects on non-small cell lung cancer patient outcomes. Int J Radiat Oncol Biol Phys. 2014;89(5):1084–91. https://doi.org/10.1016/j.ijrobp.2014.04.025.
Yellu M, Fakhrejahani F, Ying J, Mierzwa M, Malek E, Haque S, et al. Lymphopenia as a predictor of survival in chemoradiation (CRT)-treated stage III non-small cell lung cancer (NSCLC). J Clin Oncol. 2015;33(15_suppl):e18513.
Joo JH, Song SY, Park J, Choi EK, Jeong SY, Choi W. Lymphocyte depletion by radiation therapy alone is associated with poor survival in non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2016;96(2):E478.
Grossman SA, Ellsworth S, Campian J, Wild AT, Herman JM, Laheru D, et al. Survival in patients with severe lymphopenia following treatment with radiation and chemotherapy for newly diagnosed solid tumors. J Natl Compr Cancer Netw. 2015;13(10):1225–31. https://doi.org/10.6004/jnccn.2015.0151.
Fauci AS. Mechanisms of corticosteroid action on lymphocyte subpopulations. I. Redistribution of circulating T and b lymphocytes to the bone marrow. Immunology. 1975;28(4):669–80.
Nakagawa K, Garon EB, Seto T, Nishio M, Ponce Aix S, Paz-Ares L, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(12):1655–69. https://doi.org/10.1016/S1470-2045(19)30634-5.
Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(11):1454–66. https://doi.org/10.1016/S1470-2045(17)30608-3.
Mok TS, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Improvement in overall survival in a randomized study that compared Dacomitinib with Gefitinib in patients with advanced non-small-cell lung Cancer and EGFR-activating mutations. J Clin Oncol. 2018;36(22):2244–50. https://doi.org/10.1200/JCO.2018.78.7994.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–22. https://doi.org/10.1016/S1470-2045(13)70604-1.
Hida T, Nokihara H, Kondo M, Kim YH, Azuma K, Seto T, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. 2017;390(10089):29–39. https://doi.org/10.1016/S0140-6736(17)30565-2.
Camidge DR, Kim HR, Ahn MJ, Yang JC, Han JY, Lee JS, et al. Brigatinib versus Crizotinib in ALK-positive non-small-cell lung Cancer. N Engl J Med. 2018;379(21):2027–39. https://doi.org/10.1056/NEJMoa1810171.
Kim DW, Tiseo M, Ahn MJ, Reckamp KL, Hansen KH, Kim SW, et al. Brigatinib in patients with Crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung Cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490–8. https://doi.org/10.1200/JCO.2016.71.5904.
Ternyila D: Responses Observed With Poziotinib in HER2-Positive Non-Small Cell Lung Cancer. https://wwwtargetedonccom/view/responses-observed-with-poziotinib-in-her2-positive-non-small-cell-lung-cancer Date accessed as of 4/25/2021 2020.
Wang SS, Liu W, Ly D, Xu H, Qu L, Zhang L. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol Immunol. 2019;16(1):6–18. https://doi.org/10.1038/s41423-018-0027-x.
Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9(1):47. https://doi.org/10.1186/s13045-016-0277-y.
Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23(15):4242–50. https://doi.org/10.1158/1078-0432.CCR-16-3133.
Van Meter ME, McKee KY, Kohlwes RJ. Efficacy and safety of tunneled pleural catheters in adults with malignant pleural effusions: a systematic review. J Gen Intern Med. 2011;26(1):70–6. https://doi.org/10.1007/s11606-010-1472-0.
Khorrami M, Jain P, Bera K, Alilou M, Thawani R, Patil P, et al. Predicting pathologic response to neoadjuvant chemoradiation in resectable stage III non-small cell lung cancer patients using computed tomography radiomic features. Lung Cancer. 2019;135:1–9. https://doi.org/10.1016/j.lungcan.2019.06.020.
Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-fc/FcR interaction on macrophages as a mechanism for Hyperprogressive disease in non-small cell lung Cancer subsequent to PD-1/PD-L1 blockade. Clin Cancer Res. 2019;25(3):989–99. https://doi.org/10.1158/1078-0432.CCR-18-1390.
Acknowledgements
The authors would like to thank Dr. Bridget McLaughlin, technical director of the Flow Cytometry Shared Resource at UC Davis for the assistance with flow cytometry analysis.
Funding
This work was supported by the VA Merit Award (I01BX003895) (TL), Personalized Cancer Therapy Gift Fund (TL), and AstraZeneca research grant (ESR-17-12882) (TL). JZ was also supported by China Scholarship Council, grant numbers: 201906260246. The Biostatistics Shared Resource (SC) and Flow cytometry shared Resource (BML) are supported by the UC Davis Comprehensive Cancer Center Support Grant (CCSG) awarded by the National Cancer Institute (NCI P30CA093373) (SC).
Author information
Authors and Affiliations
Contributions
WM and TL contributed to the conception and design of the study. WM, CP, KY and TL contributed to the patient sample and data collection. WM, ZJ, SC, YL and TL contributed to the acquisition, analysis, or interpretation of data. WM, ZJ, SC, KT and TL drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Patient biospecimens were collected under an institutional review board (IRB)-approved protocol (Protocol No. 226210) at the University of California, Davis.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, W., Zeng, J., Chen, S. et al. Small molecule tyrosine kinase inhibitors modulated blood immune cell counts in patients with oncogene-driven NSCLC. Biomark Res 9, 69 (2021). https://doi.org/10.1186/s40364-021-00324-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00324-6