
Abstract
Background
The gut microbiota can affect neurologic disease by sha** microglia, the primary immune cell in the central nervous system (CNS). While antibiotics improve models of Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and the C9orf72 model of amyotrophic lateral sclerosis (ALS), antibiotics worsen disease progression the in SOD1G93A model of ALS. In ALS, microglia transition from a homeostatic to a neurodegenerative (MGnD) phenotype and contribute to disease pathogenesis, but whether this switch can be affected by the microbiota has not been investigated.
Results
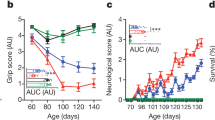
In this short report, we found that a low-dose antibiotic treatment worsened motor function and decreased survival in the SOD1 mice, which is consistent with studies using high-dose antibiotics. We also found that co-housing SOD1 mice with wildtype mice had no effect on disease progression. We investigated changes in the microbiome and found that antibiotics reduced Akkermansia and butyrate-producing bacteria, which may be beneficial in ALS, and cohousing had little effect on the microbiome. To investigate changes in CNS resident immune cells, we sorted spinal cord microglia and found that antibiotics downregulated homeostatic genes and increased neurodegenerative disease genes in SOD1 mice. Furthermore, antibiotic-induced changes in microglia preceded changes in motor function, suggesting that this may be contributing to disease progression.
Conclusions
Our findings suggest that the microbiota play a protective role in the SOD1 model of ALS by restraining MGnD microglia, which is opposite to other neurologic disease models, and sheds new light on the importance of disease-specific interactions between microbiota and microglia.
Video abstract
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by the loss of upper and lower motor neurons, leading to muscle weakness, disability, and death, with a median survival of 3 to 5 years [1]. ALS is a genetically and clinically heterogeneous disease in which the interaction between genetic background and environmental factors are thought to play a major role [1]. Familial ALS accounts for approximately 10% of cases, which results from genetic alterations in several genes including superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), chromosome 9 open reading frame 72 (C9orf72), and fused in sarcoma (FUS) [2]. The remaining 90% of cases are sporadic, suggesting an important environmental component.
The intestinal microbiota encompasses trillions of organisms that inhabit the gut [3] and can play a role in neurologic diseases by modulating immune responses in the CNS, altering endocrine signaling along the hypothalamic pituitary axis, and by directly signaling through afferent nerves [4]. Studies have found alterations in the gut microbiota of patients with ALS. Brenner et al. studied 25 patients with ALS vs. 32 healthy controls [5] and found alterations in Ruminococcaceae. Blacher et al. analyzed a cohort of 37 patients with ALS vs. 29 healthy controls and found decreased abundance of microbial genes involved in nicotinamide and tryptophan metabolism [6]. Nicholson et al. sequenced the largest number of ALS subjects to date (n = 68) vs. healthy controls (n = 61) and found a decrease in the butyrate producing bacteria, Roseburia intestinalis and Eubacterium rectale [7]. The changes in butyrate producing bacteria are consistent with a case study in which 5 patients with ALS had low levels of other butyrate-producing bacteria [4) compared to unique genes (Supplemental Figs. 5 and 6). Antibiotics modulated 27 genes in the same direction in both WT and SOD1 mice (14 up and 13 down), including upregulation of the MGnD regulator Apoe and downregulation of ubiquitin binding protein (Ubc), involved in autophagy. Antibiotics modulated some genes in opposing directions in WT and SOD mice, including Tmem175, a lysosomal potassium channel which plays an important role in clearance of autophagosomes [56], and Gpr165, a homeostatic microglia gene. These findings indicate that the microbiota may have opposing effects on microglial function in healthy vs. diseased animals.
Discussion
Our study confirms and extends the finding that the gut microbiota plays a protective role in the SOD1 model of ALS and identifies a new mechanism related to microglia. The microbiota influence over microglia could be mediated by modulating peripheral immune cells that traffic to the CNS or the production of metabolites, which warrants further study. Microglia play an important role in the maintenance of brain homeostasis but lose this homeostatic function in ALS [20, 57]. We recently identified a neurodegenerative molecular signature in microglia from SOD1 mice which we termed MGnD, in contrast to a homeostatic microglial phenotype [16]. Studies have shown that the gut microbiota maintain microglial function in homeostasis and that antibiotics can decrease homeostatic microglia signatures [27]. Consistent with this, we found that antibiotics decreased homeostatic genes P2ry12, P2ry13, and Cst3 in SOD1 mice. However, we did not observe a decrease in homeostatic genes in antibiotic treated WT mice, as previously reported in Erny et al. [27]. A key difference is that we used a low-dose antibiotic treatment regimen that modulated the microbiota, whereas Erny et al. used a high-dose antibiotic treatment regimen that mimicked a germ-free state. Unique to our study, we found that antibiotics increased MGnD genes in SOD1 mice, including Apoe, Cst7, Lgals, Axl, and others, which is opposite to the effects observed in WT mice and models of AD and PD [11, 27, 28]. We found 109 genes modulated by antibiotics that enhanced SOD1 vs. WT changes in microglia, indicating that an antibiotic-induced dysbiosis amplifies a microglia MGnD phenotype. Furthermore, we found that changes in microglia preceded altered motor dysfunction in antibiotic-treated mice, suggesting that the microbiota slows disease progression by restraining a neurodegenerative microglial phenotype. It is possible that some of these genes, including granulin, reflect early markers of progression and may be a repair response to neurodegeneration. Grn is linked with disease progression in ALS as it is elevated in patients and mice that have progressed in their disease course but is not elevated at disease onset ALS patients compared to healthy controls or pre-symptomatic SOD1 mice [53]. While elevated Grn is associated with ALS, it is suggested to play a protective role by restraining microglia inflammation that leads to neurotoxicty [55].
Because the SOD1 model exhibits a progressive disease that requires more than 4 months of antibiotic intervention, we utilized two microbiota interventions that could lead to a chronic mild depletion (antibiotics) or augmentation (co-housing). In our study, we selected a low-dose antibiotic regimen shown to be well tolerated for long-term administration [10]. This leads to antibiotic-induced dysbiosis, rather than full microbiota depletion. We found that antibiotics initially depleted most populations of the endogenous microbiota, then led to an increase in antibiotic resistant organisms, which may be linked to the altered disease progression that we observe later in the disease course. Antibiotic treatment did not affect the time of disease onset but did affect disease progression, potentially suggesting that administration of antibiotics in the early-symptomatic phase could also have a similar effect of the disease course.
We identified several groups of bacteria depleted by antibiotics that may have beneficial roles in ALS. The Gram-negative anaerobe, Akkermansia was depleted at multiple time points. Akkermansia has recently been shown to ameliorate disease in antibiotic pre-treated SOD1 mice, which is linked to the production of nicotinomide [58]. Akkermansia may also have beneficial roles for other neurologic diseases, including multiple sclerosis [43], epilepsy [59], and Alzheimer’s disease [60]. Antibiotics also depleted several members of Clostridial clusters IV and XIVa, which are major butyrate producers in the gut. Two independent studies have found that butyrate producing bacteria were depleted in ALS [7, 15]. We did not observe motor dysfunction in antibiotic-treated non-transgenic WT mice. Thus, based on our experimental data, it appears that low-dose antibiotics worsen disease only in genetically susceptibly hosts.
It is possible that antibiotics could have an off-target effect and that antibiotic-mediated toxicity may be responsible for the changes, rather than antibiotic-induced alterations in the gut microbiota. A higher dose combination of antibiotics in the C9orf72 model of ALS initially reduced microglia infiltration into the CNS and ameliorated disease, but then led to off-target health consequences [29]. We did not observe a MGnD microglial phenotype in low-dose antibiotic-treated WT mice. Furthermore, in an animal model of AD, this low-dose combination of 8 antibiotics was well tolerated for several months and reversed a MGnD microglial phenotype [10, 28].
Several studies have found that co-housing a genetically susceptible mouse with a wildtype mouse can transfer disease phenotype, with either the pathogenic or protective trait transferred by co-housing [31,32,33,34]. For example, cohousing 5XFAD and WT mice led to cognitive impairment in the WT mice, which was associated with infiltration of Th1 cells in the brain and increased inflammatory cytokines [31]. In our co-housing experiment, we saw no effect on motor function, neurologic score, or survival in SOD1 or WT mice, suggesting that this trait is not transmissible via the microbiota alone. Cohousing may have less of an impact than antibiotics on the microbiome due to inherent colonization resistance. Furthermore, an important point for our study is that we also saw little difference between SOD1 mice and WT littermate microbiota. Other studies have found that differences in the gut microbiota in WT vs SOD1 mice are vivarium dependent [6], and vivarium-dependent changes in the gut microbiota in the C9orf72 ALS animal can determine disease susceptibility [29]. Thus, in a colony of SOD1 mice that have a microbiota distinct from WT mice, cohousing could alter potentially alter disease progression.
Conclusions
In summary, our study highlights the importance of disease-specific interactions between the microbiota and microglia. Furthermore, given the critical role of microglia in ALS, our finding that the microbiota restrains neurodegenerative microglia in SOD1 mice has important implications for the pathogenesis and treatment of subjects with ALS as detrimental effects of antibiotics have been observed in ALS patients. We identified two groups of bacteria that have been reported to have beneficial roles in ALS, including Akkermansia and butyrate-producing bacteria, and further work is needed to confirm their protective role. Finally, we were not able to confer disease protection to SOD1 mice or transmit disease to WT littermates via cohousing, highlighting the importance of the interaction of genetic and environmental risk in the SOD1 model of ALS.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI Short Read Archive data repository, under NCBI Bioproject number PRJNA769453.
References
Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–28. https://doi.org/10.1038/nrneurol.2013.203.
Stephenson J, Amor S. Modelling amyotrophic lateral sclerosis in mice. Drug Discov Today Dis Models. 2017;25-26:35–44. https://doi.org/10.1016/j.ddmod.2018.10.001.
Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. 2016;375:2369–79. https://doi.org/10.1056/NEJMra1600266.
Cox LM, Weiner HL. Microbiota signaling pathways that influence neurologic disease. Neurotherapeutics. 2018;15:135–45. https://doi.org/10.1007/s13311-017-0598-8.
Brenner D, et al. The fecal microbiome of ALS patients. Neurobiol Aging. 2018;61:132–7. https://doi.org/10.1016/j.neurobiolaging.2017.09.023.
Blacher E, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature. 2019;572:474–80. https://doi.org/10.1038/s41586-019-1443-5.
Nicholson K, et al. The human gut microbiota in people with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:186–94. https://doi.org/10.1080/21678421.2020.1828475.
Rowin J, **a Y, Jung B, Sun J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol Rep. 2017;5. https://doi.org/10.14814/phy2.13443.
Berer K, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–41. https://doi.org/10.1038/nature10554.
Minter MR, et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of Alzheimer's disease. Sci Rep. 2017;7:10411. https://doi.org/10.1038/s41598-017-11047-w.
Sampson TR, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469–1480.e1412. https://doi.org/10.1016/j.cell.2016.11.018.
Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4615–22. https://doi.org/10.1073/pnas.1000082107.
Miller PG, Bonn MB, Franklin CL, Ericsson AC, McKarns SC. TNFR2 deficiency acts in concert with gut microbiota to precipitate spontaneous sex-biased central nervous system demyelinating autoimmune disease. J Immunol. 2015;195:4668–84. https://doi.org/10.4049/jimmunol.1501664.
Ochoa-Reparaz J, Mielcarz DW, Haque-Begum S, Kasper LH. Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes. 2010;1:103–8. https://doi.org/10.4161/gmic.1.2.11515.
Sun J, et al. Antibiotics use and risk of amyotrophic lateral sclerosis in Sweden. Eur J Neurol. 2019;26:1355–61. https://doi.org/10.1111/ene.13986.
Krasemann S, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581.e569. https://doi.org/10.1016/j.immuni.2017.08.008.
Appel SH, Zhao W, Beers DR, Henkel JS. The microglial-motoneuron dialogue in ALS. Acta Myol. 2011;30:4–8.
Boillee S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–92.
Brettschneider J, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7:e39216. https://doi.org/10.1371/journal.pone.0039216.
Butovsky O, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77:75–99. https://doi.org/10.1002/ana.24304.
Butovsky O, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122:3063–87. https://doi.org/10.1172/JCI62636.
Turner MR, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–9. https://doi.org/10.1016/j.nbd.2003.12.012.
Henkel JS, et al. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–35.
Kuhle J, et al. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur J Neurol. 2009;16:771–4. https://doi.org/10.1111/j.1468-1331.2009.02560.x.
Ryberg H, et al. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve. 2010;42:104–11. https://doi.org/10.1002/mus.21683.
Yamanaka K, et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A. 2008;105:7594–9.
Erny D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–77. https://doi.org/10.1038/nn.4030.
Dodiya HB, et al. Sex-specific effects of microbiome perturbations on cerebral Abeta amyloidosis and microglia phenotypes. J Exp Med. 2019;216:1542–60. https://doi.org/10.1084/jem.20182386.
Burberry A, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 2020;582:89–94. https://doi.org/10.1038/s41586-020-2288-7.
Minter MR, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep. 2016;6:30028. https://doi.org/10.1038/srep30028.
Wang X, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29:787–803. https://doi.org/10.1038/s41422-019-0216-x.
Benakis C, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. 2016;22:516–23. https://doi.org/10.1038/nm.4068.
Regen T, et al. IL-17 controls central nervous system autoimmunity through the intestinal microbiome. Sci Immunol. 2021;6. https://doi.org/10.1126/sciimmunol.aaz6563.
Zhang Y, et al. Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome. 2019;7:116. https://doi.org/10.1186/s40168-019-0733-3.
Scott S, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4–15. https://doi.org/10.1080/17482960701856300.
Hatzipetros T, et al. A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J Vis Exp. 2015. https://doi.org/10.3791/53257.
Butovsky O, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–43. https://doi.org/10.1038/nn.3599.
Richner M, Jager SB, Siupka P, Vaegter CB. Hydraulic extrusion of the spinal cord and isolation of dorsal root ganglia in rodents. J Vis Exp. 2017;55226. https://doi.org/10.3791/55226.
Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–8. https://doi.org/10.1038/nmeth.2639.
Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–78. https://doi.org/10.1038/nprot.2012.016.
Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–30. https://doi.org/10.1093/bioinformatics/btt703.
Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. https://doi.org/10.1038/ismej.2012.8.
Cox LM, et al. Gut microbiome in progressive multiple sclerosis. Ann Neurol. 2021;89:1195–211. https://doi.org/10.1002/ana.26084.
Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. https://doi.org/10.1038/nmeth.f.303.
Yilmaz P, et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42:D643–8. https://doi.org/10.1093/nar/gkt1209.
Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. https://doi.org/10.1186/gb-2011-12-6-r60.
Bokulich NA, et al. q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. mSystems. 2018;3. https://doi.org/10.1128/mSystems.00219-18.
Cox LM, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–21. https://doi.org/10.1016/j.cell.2014.05.052.
Abriouel H, et al. The controversial nature of the Weissella genus: technological and functional aspects versus whole genome analysis-based pathogenic potential for their application in food and health. Front Microbiol. 2015;6. https://doi.org/10.3389/fmicb.2015.01197.
Perez Visñuk D, Savoy de Giori G, LeBlanc JG, de Moreno de LeBlanc A. Neuroprotective effects associated with immune modulation by selected lactic acid bacteria in a Parkinson’s disease model. Nutrition. 2020;79-80:110995. https://doi.org/10.1016/j.nut.2020.110995.
Gres V, Kolter J, Erny D, Henneke P. The role of CNS macrophages in streptococcal meningoencephalitis. J Leukoc Biol. 2019;106:209–18. https://doi.org/10.1002/jlb.4mr1118-419r.
Vasek MJ, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534:538–43. https://doi.org/10.1038/nature18283.
Philips T, et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J Neuropathol Exp Neurol. 2010;69:1191–200. https://doi.org/10.1097/NEN.0b013e3181fc9aea.
Herdewyn S, De Muynck L, Van Den Bosch L, Robberecht W, Van Damme P. Progranulin does not affect motor neuron degeneration in mutant SOD1 mice and rats. Neurobiol Aging. 2013;34:2302–3. https://doi.org/10.1016/j.neurobiolaging.2013.03.027.
Martens LH, et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–9. https://doi.org/10.1172/JCI63113.
**n S, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A. 2017;114:2389–94. https://doi.org/10.1073/pnas.1616332114.
Chiu IM, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4:385–401. https://doi.org/10.1016/j.celrep.2013.06.018.
Blacher E, Levy M, Tatirovsky E, Elinav E. Microbiome-modulated metabolites at the interface of host immunity. J Immunol. 2017;198:572–80. https://doi.org/10.4049/jimmunol.1601247.
Olson CA, et al. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell. 2018;174:497. https://doi.org/10.1016/j.cell.2018.06.051.
Ou Z, et al. Protective effects of Akkermansia muciniphila on cognitive deficits and amyloid pathology in a mouse model of Alzheimer’s disease. Nutr Diabetes. 2020;10:12. https://doi.org/10.1038/s41387-020-0115-8.
Zhang YG, et al. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin Ther. 2017;39:322–36. https://doi.org/10.1016/j.clinthera.2016.12.014.
Berard M, et al. Ralstonia pickettii-induced ataxia in immunodeficient mice. Comp Med. 2009;59:187–91.
Gordon PH, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–53. https://doi.org/10.1016/S1474-4422(07)70270-3.
Acknowledgements
Microglial cells were sorted at the flow cytometry core facility at the Ann Romney Center for Neurologic Diseases. Microbiota samples were sequenced at the Harvard Biopolymers facility, and microglia were sequenced at the Broad Institute.
Funding
This work was supported by grants from the NIH/NINDS 1R01NS115951 (H.L.W), by the Brigham Research Institute NextGen Award (L.M.C), NIH-NINDS (R01NS088137) (O.B.), and NIH-NIA (R01AG051812, R01AG054672) (O.B.).
Author information
Authors and Affiliations
Contributions
L.M.C, N.C., O.B., and HLW designed the study and interpreted data. N.C. administered treatments to SOD1 mice, performed neurologic scoring, and led experiments to sort microglia from the spinal cord. L.M.C sequenced and analyzed the gut microbiota and contributed to the analysis of behavioral and microglia data. CM and CG analyzed microglia transcriptional profiles and performed pathway analysis. The authors contributed to the writing of the manuscript. The authors read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal studies were carried out in accordance with IACUC-approved protocols.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplemental Figure 1.
Effect of antibiotics on weight in wild-type littermate mice. WT littermate mice were treated with antibiotics (ABX), co-housed with SOD1 mice (CoHo), or untreated (H2O), n = 11-12 per group and change in weight and motor function was assessed until SOD1 mice in each treatment group reached humane endpoint criteria (Fig. 1). a) Weight was increased in antibiotic treated mice. Data represent mean ± standard error of the mean (SEM). Multiple unpaired t-tests, adjusted for false-discovery rate, * q< 0.05. b) Neurologic score was assessed along with SOD1 mice. No motor deficits were observed. c) Mice were trained on the rotarod until they could maintain balance for 285 seconds. WT mice were tested along SOD1 mice. No loss in motor function was observed. b-c) data shifted up slightly so that all three groups can be seen.
Additional file 2: Supplemental Figure 2.
Disease progression and microbiota changes in a second cohort of mice. A second cohort of SOD1 and WT mice were treated with antibiotics, cohoused, or did not receive treatment (H2O) n = 6 per group. Microglia were then sorted at day 120 of life. a) Neurologic scores were measured 3 times a week, and no difference between treatment groups was observed prior to day 120, which is similar to effects observed in the survival cohort (Fig. 1). b-d) Microbiota samples were collected at day 30 of life (baseline), then at days 37, 51, and 93. b) ADONIS test of microbiota samples from both the survival and microglia sort cohort indicates that treatment and timepoint have a substantial contribution to microbiome variation, whereas the contribution of cage and cohort is small, and genotype has no effect. *** p =0.001 c) Principal coordinates analysis of unweighted UniFrac distances show that samples cluster at baseline, and are shifted by antibiotic treatment, but not genotype or cohousing. d) Microbiota composition over time shows expansion of Ralstonia, Streptococcus, and Weisella, which is similar microbiota changes observed in the survival cohort (Fig. 2).
Additional file 3: Supplemental Figure 3.
The effect of genotype on microglia gene expression in untreated and antibiotic treated SOD1 and WT mice. a) Differential genes (SOD1 vs WT) in both untreated and antibiotic treated mice. DESeq FDR-adjusted q value < 0.2. b) The top 50 upregulated genes up-regulated and down-regulated in both treated and untreated mice. c) DESeq2 normalized levels of selected genes consistently down- or up-regulated by genotype. * p < 0.05, ** p < 0.01, *** p < 0.001.
Additional file 4: Supplemental Figure 4.
Unique genes modulated by antibiotics in SOD1 mice. A) Differential genes in SOD1-antibiotic (ABX) vs SOD1-H2O that are not altered in SOD1-H2O vs. WT-H2O (Fig. 3). DESeq FDR-adjusted q value < 0.2.
Additional file 5: Supplemental Figure 5.
Unique genes modulated by antibiotics in WT mice. a) Top 50 genes up and down regulated by antibiotics uniquely in WT mice. DESeq FDR-adjusted q value < 0.2.
Additional file 6: Supplemental Figure 6.
Genes altered by antibiotics in both SOD1 and WT mice. A-B) Genes modulated by antibiotics in Wt (a) or SOD1 (b). Several genes are regulated in the same direction (upper plot) while others are differentially regulated by antibiotics according to genotype (lower plot). DESeq FDR adjusted q value < 0.2. c) Representative genes modulated by antibiotics.
Additional file 7: Supplementary Table 1.
Longitudinal comparison of changes in unweighted UniFrac distances.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cox, L.M., Calcagno, N., Gauthier, C. et al. The microbiota restrains neurodegenerative microglia in a model of amyotrophic lateral sclerosis. Microbiome 10, 47 (2022). https://doi.org/10.1186/s40168-022-01232-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-022-01232-z