
Abstract
Background
A motor unit (MU) is formed by a single alpha motor neuron (MN) and the muscle fibers it innervates. The MU is essential for all voluntary movements. Functional deficits in the MU result in neuromuscular disorders (NMDs). The pathological mechanisms underlying most NMDs remain poorly understood, in part due to the lack of in vitro models that can comprehensively recapitulate multistage intercellular interactions and physiological function of the MU.
Results
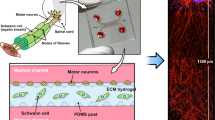
We have designed a novel three-dimensional (3D) bilayer hydrogel tri-culture system where architecturally organized MUs can form in vitro. A sequential co-culture procedure using the three cell types of a MU, MN, myoblast, and Schwann cell was designed to construct a co-differentiating tri-culture on a bilayer hydrogel matrix. We utilized a µ-molded hydrogel with an additional Matrigel layer to form the bilayer hydrogel device. The µ-molded hydrogel layer provides the topological cues for myoblast differentiation. The Matrigel layer, with embedded Schwann cells, not only separates the MNs from myoblasts but also provides a proper micro-environment for MU development. The completed model shows key MU features including an organized MU structure, myelinated nerves, aligned myotubes innervated on clustered neuromuscular junctions (NMJs), MN-driven myotube contractions, and increases in cytosolic Ca2+ upon stimulation.
Conclusions
This organized and functional in vitro MU model provides an opportunity to study pathological events involved in NMDs and peripheral neuropathies, and can serve as a platform for physiological and pharmacological studies such as modeling and drug screening. Technically, the rational of this 3D bilayer hydrogel co-culture system exploits multiple distinct properties of hydrogels, facilitating effective and efficient co-culturing of diverse cell types for tissue engineering.
Similar content being viewed by others

Background
The Neuromuscular system includes motor neurons (MNs) and their innervated skeletal muscles [26], we employed a simpler (without Schwann cells and Matrigel) in vitro MU model containing only MNs and myotubes on µ-molded hydrogel to study NMJ formation. Using this earlier system, we have successfully demonstrated that the expression level of a retinoic acid signaling protein, Cellular Retinoic Acid Binding Protein 1 (CRABP1) in MNs affected the length of axons and AChR cluster formation. The requirement for CRABP1 in MU functionality in vivo was validated in Crabp1 knockout mice and confirmed using gene rescue experiments. Therefore, this 3D MU system can be a powerful tool to efficiently identify molecular players and dissect signaling pathways critical to formation and function of MUs or NMJs. For a practical consideration (time and effort), this system provides a reliable and efficient MU model for numerous studies including physiological and pharmacological investigations.
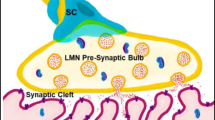
The formation of functional NMJs in the 3D tri-culture system is also supported by data shown in our previous study [26] where a similar, but simpler (only two cell types, MN and C2C12), version of co-culture system was reported. This earlier study has detected the expression of pre-synaptic vesicle protein SV2 and post-synaptic AChR in NMJs [26]. The expression of synaptic vesicle glycoprotein-2 would indicate the ability of MNs to release neurotransmitter from the axon terminals. To improve the physiological relevance of this earlier model, in this current study, we added the Matrigel layer embedded with a third cell type important for NMJ, i.e. Schwann cells. This has apparently improved this model as demonstrated in multiple measurements including extended axon length, myelinated axons, and axon terminals. In the NMJ of this tri-culture model (Fig. 4a), the presence of two groups of small branches of the axon terminal (White and blue arrows, left panel) would suggest that the MN can innervate multiple myotubes. The morphology of small branches would indicate the potential of NMJ to form a pretzel-shaped structure, a classic morphological feature of NMJs as shown in murine models (Fig. 4a, left panel). In the post-synaptic compartment, the expression of AChR is clearly detected, predominantly near the MN especially at its axon terminal (Fig. 4a, Additional file 1: Supplementary data, Fig S2), indicating that the MN recruits AChRs to its axon terminal to form AChR clusters. The functionality of NMJ is demonstrated using a conventional live imaging method that detects the dynamics of Ca2 + reflecting the functional response of neurons (MN in this case) and muscles (myotubes in this case). Ca2 + imaging data validate the relevant responses of key components in the MUs—increases in intracellular Ca2 + upon high potassium (KCl) or acetylcholine stimulation for MNs (electrophysiological response), or myotubes (contractility), respectively, but not in Schwann cells as predicted. Therefore, each cellular component (MNs, myotubes, and Schwan cells) appropriately exhibits its biological response (or lack thereof) to stimuli, supporting the physiological relevance of this improved, tri-culture model. As shown in Fig. 4b and Additional file 3: video S2, both myotube contraction and the maximum displacement occurred specifically at the axon terminal. Additionally, the contraction frequency of the MN-driven myotubes was fivefold greater than that of the spontaneous contraction of myotubes without MNs nearby (Fig. 4c and Additional file 3: video S2). Taken together, these results support the formation of functional NMJs in the 3D tri-culture.
We recognize that there remain limitations in the application of this in vitro 3D culture MU model. The purpose of using cell lines to generate this MU model is for the sake of consistency, ease in culture procedure and timesaving. It remains to be determined whether appropriate human cell lines, or stem cell systems, such as iPSC differentiated MNs or muscle cells, can be similarly optimized for this system [14]. Secondly, in Ca2+ imaging experiments, due to the thickness of Matrigel layer, it will be challenging to collect clear signals from both MNs and myotubes at the same time because the cells reside in different layers (Fig. 5a). This would limit real-time assessment of MN excitation together with MN-driven myotube contraction. However, advanced imaging tools may be useful to improve temporal resolution of the assay. While the percentage of MNs and myofibers that responded to physiological stimuli with an increase in [Ca2+] was quite high, the amplitude of the responses was variable. Possible explanations for this variability likely result from the challenges of accurately correcting background fluorescence in a 3D population of cells and variable drug application and dye loading through the layers of the gel matrix. These issues might be addressed in future experiments by using genetically encoded Ca2+ indicators targeted to specific cell types and local drug application by micropipette. Overall, the Ca2+ recordings described here demonstrate the functional viability of this new model system.
It is worthy to note the well differentiated and organized mature myotubes in this 3D bilayer hydrogel tri-culture system. The mature myotubes are similar to skeletal muscle fibers [27], including their morphology, similar width, length and a cylindrical, aligned, striated, and unbranched shape. The µ-molded hydrogel contributes to the alignment of myotubes and the Matrigel may provide the extracellular matrix-like microenvironment to improve myotube maturation. Additionally, the Matrigel also protects the contracting myotubes, preventing them from detachment in a long-tern culture.
Conclusion
We report a procedure to engineer a structurally organized in vitro 3D MU model that can be reliably generated within a reasonably short period of time. This MU model is validated with regards to morphology, essential biomarkers and the function of a MU including contractility and calcium signaling. Three key advantages are noted in this new model. First, the entire MU (single MN) can be examined, and subcellular/functional details can be studied. Secondly, the included Schwann cells allows proper myelination to occur, enabling detailed studies of MN regulation with regards to NMJ development/maturation and maintenance. Thirdly, using this system, it is possible to generate various in vitro disease models by manipulating each relevant cell type. Thus, this new system is appropriate not only for basic research, but also for disease modeling and drug screening.
Methods and materials
Cell lines and its culture condition
The mouse C2C12 myoblast cell line was purchase from ATCC (Cat. no. CRL-1772). The mouse motor neuron cell line MN1 were kindly provided by Ahmet Hoke (The Johns Hopkins University, Baltimore, MD). The mouse Schwann cell line IMS32 were purchased from CosmoBio Co., LTD (Cat no. PMC-SWN-IMS32C). The three Cell lines were maintained in high glucose Dulbecco’s Modified Eagle Medium (DMEM) with 2 mM L-glutamine (Thermo Fisher Scientific, cat. no. 11965–092) supplemented with 10% FBS (ALTANTA Biologics, cat. no. S11150), 100 U/mL penicillin and 100 mg/mL streptomycin (Thermo Fisher Scientific, cat. no. 15140–122). Cells were maintained at 37 °C in a humidified 5% CO2 cell culture incubator. The C2C12 Cells were sub-cultured using 0.25% trypsin (Thermo Fisher Scientific, cat. no. 25200–056) upon reaching approximately 70% confluence. The MN1 cells were sub-cultured using 0.05% trypsin (Thermo Fisher Scientific, cat. no. 25200–054) upon reaching approximately 90% confluence. The IMS32 cells were sub-cultured using 5 mM EDTA (Thermo Fisher Scientific, cat. no. 15575–020) upon reaching approximately 70–80% confluence.
The procedure for the 3D bilayer hydrogel tri-culture system is described in Fig. 1b
Stamp fabrication
PDMS stamps were fabricated as described elsewhere [24, 26, 28]. Briefly, the silicon wafer mold was treated with trichloromethyl silane (Millipore Sigma, cat. no. 75-79-6) vapor for 30 min as a releasing agent. PDMS mixture (PDMS: curing agent = 10:1, Sylgard 184, Dow Corning) was poured onto the master mold and cured for 2 h. at 60 °C. The cured PDMS was excised from the master mold and trimmed with a clean razor blade to generate the stamp. The PDMS stamp was then cleaned with oxygen plasma (irradiation intensity: 200 W) for 10 min.
Micro-molded (µ-molded) hydrogel fabrication
Under sterile conditions, prepare a 10% solution of gelatin A (Sigma, cat. no. G1890-100 g) was prepared in sterile ddH2O. The solution was placed on a rotating rack at 65 °C for 10 min or until thoroughly dissolved, then removed from rotation to allow bubbles to rise out of the solution for 10 min. A 65 °C hotplate (Apollo; Digital Heating and Chilling plate) was used to create the µ-molded hydrogel and adhere it to the cover glass (Fisher Scientific, cat. no. 1254580), so µ-mold PDMS stamps, gelatin, and coverslip glass were warmed to 65 °C before molding. 18 µl of gelatin solution was carefully pipetted directly onto the stamp, then quickly place the cover glass on top of the gelatin droplet ensuring full coverage of the glass. The stamps were remove from the hotplate and the entire sandwich was allowed to cool at room temperature for 30 min. Under sterile conditions, the cover glass and hydrogel was carefully peeled off from the stamp using forceps and placed in a 24 well plate. The hydrogels were exposed to UV for 5 min.
The gelatin hydrogel was crosslinked with a 5% transglutaminase solution for 18 h. 1 g of powdered transglutaminase was mixed with 19 ml of autoclaved ddH2O under sterile conditions. The enzyme was placed on a rotating rack at 50 °C for 10 min to dissolve and allowed to cool at room temperature before filtration. The transglutaminase solution was filtered through a 0.2 µm filter. After hydrogels have had 5 min under UV, they were ready for the addition of transglutaminase. Leave the 24 well plate covered in the hood at room temperature for 18 h. After 18 h, transglutaminase was suctioned out. The hydrogel was then rinsed three times by adding autoclaved ddH2O to the well. On the last rinse, the 24 well was placed under UV uncovered for 5 min. Following UV exposure, the 24 well plate can be covered, placed in a sterile bag, and stored at 4 °C until use.
MN1 transfection
Plasmid CLYBL-(Ef1a-SBP-LNGFR-T2A-mApple)-(CAG-rtTA)-(TRE-hNIL)) was purchased from Addgene (cat. no. 05842). MN1 cells transfection was performed by Lipofectamine 3000 (Thermo Fisher Scientific, cat. no. L3000-015) following the manufacturer’s instruction. MN1 cells were added to the tri-culture system 1 dayafter transfection.
Cell culture in the system
On day 0, 2 × 104 C2C12 cells/cm2 were seeded on hydrogels in 24-well plates and cultured in growth medium. Once confluent (about 2 day), myotube differentiation medium (DMEM with 2% horse serum (Thermo Fisher Scientific, cat. no. 5H30074.2), 100 U/mL penicillin, 100 mg/mL streptomycin and 2 mM L-glutamine) was added. C2C12 were differentiated for 4 days and the myotube differentiation medium was replenished every 2 days.
On Day 6, Schwann cell line IMS32 was trypsinized by 0.05% trypsin and re-suspended in cold myotube differentiation medium in the density of 4 × 104 cells/ml. Schwann cells were mixed with Matrigel in a 1-to-1 ratio and then kept on ice. Matrigel Matrix (Corning, cat. no. 356255) was thaw in ice in cold room over night before use. The medium in the system was removed and 18 µl of the mixture was added on the center of the hydrogel. The system was kept in 37 °C incubator for 3 min, and added the fresh myotube differentiation medium.
On day 7, the transfected 1 × 103 MN1 cells/cm2 were added to each well of 24-well, on top of the differentiated C2C12 cells. MN1 cells were previously transfected with either empty vector control or GFP-Crabp1 prior to seeding onto C2C12 cells. The co-culture was maintained in MN differentiation medium 1 (DMEM media with 100 U/mL penicillin, 100 mg/mL streptomycin, 2 mM L-glutamine, 1% MEM non-essential amino acids (NEAA; Thermo Fisher Scientific, cat. no. 11140–050), 2 µg/ml doxycycline (Sigma, cat. no. D9891) and 10 um Y27632 Dihydrochloride (R&D, cat no. 1254/1) for 4 days, then changed to MN differentiation medium 2 (MN differentiation medium 1 without Y27632 Dihydrochloride). The MN differentiation mediums was replenished every two days.
Immunostaining
For Immunostaining, the tri-culture slides were fixed in 100% methanol for 5 min at – 20 °C. After three times of 5 min 1XPBS wash, 2% BSA was added for 1 h at room temperature for blocking. Primary antibodies, α-actinin (1:100; Cell Signaling, cat. no. 6487S), Tuj1 (1:1000; R and D, cat. no. MAB 1196) or myelin basic protein MBP (1:500; R and D, Cat. No. MAB42282) in 1XPBS were incubated overnight at 4 °C. After three times of 5 min 1XPBS wash, fluorescent secondary antibodies (1:1000), and µBTX conjugated to a fluorescent dye (1:400; Biotium, Cat No. 00005) were incubated for 2 h in dark at room temperature. The DAPI (1:1000; Thermal Fisher Scientific, Cat. No. 62248) in 1 × PBS were incubated for 10 min in dark at room temperature. The slide then was washed three times of 5 min 1xPBS. Z-stack images were captured on Olympus Fluoview FV1000 BX2 Upright Confocal.
Calcium imaging
Tri-cultures on 12.5 mm glass coverslips were briefly washed in HEPES Hanks’ Buffer Solution (HHBS) (5.6 mM glucose, 20 mM Hepes, 3 mM NaHCO3, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 137 mM NaCl, 5.0 mM KCl, 4.9 mM MgCl2, 4.1 mM MgSO4, 1.26 mM CaCl2, pH = 7.4) and then the coverslip was incubated with fura-2 AM in 0.04% pluronic acid dissolved in HHBS to a final concentration of 20 μM for 30 min at 37 °C. After loading indicator, coverslips were washed in fresh HBSS for 10 min at 37 °C and then transferred to an imaging chamber for microscopy experiments.
Fura-2 imaging was performed on an Olympus IX71 microscope using a 20X objective lens. Excitation wavelength was selected with a galvanometer-driven monochromator (8 nm slit width) coupled to a 75 W xenon arc lamp (Optoscan; Cairn Research). Intracellular Ca2 + concentration ([Ca2 +]i) was monitored by sequential excitation (1 Hz) of fura-2 at 340 and 380 nm. A single image was acquired at 610 nm to identify mApple expressing MNs in the field. Fluorescent images were captured using a cooled charge-coupled device camera (Cascade 512B; Roper Scientific) and Metafluor acquisition software (Molecular Devices). All fura-2 experiments were performed in HBSS buffer at room temperature. To depolarize MNs the extracellular K + concentration was increased to 50 mM with K + exchanged reciprocally for Na + in HHBS. For myotube stimulation 10 μM ACh was applied by bath perfusion. A 60 s baseline in HHBS was acquired before stimulation of each coverslip. High K + was applied for 30 s and ACh for 1 min. After stimulus application, a washout of 60 s was performed. Background images were acquired after each experiment; the coverslip was wiped clean, then 20 frames were acquired using the same parameters as above.
Calcium imaging data analysis
Images were processed using MetaFluor Analyst to draw regions of interest (ROIs), subtract background, and generate 340/380 ratios. MN ROIs were selected based on mApple expression and further verified by morphology by a blinded expert. Differentiated MNs each has a cell body and a single enlarged axon terminal. Myotube ROIs were selected based on morphological criteria; myotubes exhibit a distinct elongated, skeletal muscle fiber-like morphology. Given that myotubes can span the total length of the field, three ROIs for each individual myotube were drawn and averaged to represent a single cell.
Peak amplitude analysis was performed by comparing the average baseline [Ca2+]i to the maximum reached after stimulation. Only responding cells were included in peak amplitude analysis. A response was defined by the following criteria: (1) a signal intensity of at least 450 (16-bit scale) after background subtraction, and (2) a peak [Ca2+]i at least two standard deviations (SD) above baseline (60 s average). Criterion “1” was empirically determined to exclude cells with poor signal to noise ratio. [Ca2+]i calculations were performed in Microsoft Excel by averaging 340/380 intensity ratios for MNs or myotubes in the field that responded to stimulation.
Statistical analysis
Data were analyzed using the Student’s t test. Statistical analyses were performed using Prism 8.0 (GraphPad, CA). All tests were performed at a significance level of p ≤ 0.05, and data are presented as mean ± standard error or mean ± SD as indicated. For statistical analyses of calcium image, MN and myotube data were assessed for normality using the D'Agostino-Pearson normality test. Myotube data exhibited normality, whereas MN did not. Therefore, myotube data was subjected to paired Student’s t-test and MN data were subjected to the non-parametric Wilcoxon signed-rank test8. For each experiment “n” was defined by a single microscopic field on a single coverslip. Percent responders was calculated using number of responders/total MNs or myotubes in the field.
Availability of data and materials
All data are available upon request.
Abbreviations
- MU:
-
Motor unit
- MN:
-
Motor neuron
- NMD:
-
Motor neuron disorder
- 3D:
-
Three-dimensional
- NMJ:
-
Neuromuscular junction
- Ach:
-
Acetylcholine
- ESC:
-
Embryonic stem cell
- Ipsc:
-
Induced pluripotent stem cell
- µ-molded:
-
Micro-molded
- NGN2:
-
Neurogenin 2
- ISL1:
-
ISL LIM Homeobox 1
- LHX3:
-
LIM Homeobox 3
- MBP:
-
Myelin basic protein
- AChR:
-
Acetylcholine receptor
References
Li L, **ong WC, Mei L. Neuromuscular junction formation, aging, and disorders. Annu Rev Physiol. 2018;80:159–88.
Eccles JCSC. Numbers and contraction values of individual motor units examined in some muscles of the limb. Proc R Soc Lond B Biol Sci. 1930;106:326–57.
Nishimune H. Active zones of mammalian neuromuscular junctions: formation, density, and aging. Ann N Y Acad Sci. 2012;1274(1):24–32.
Koczwara KE, Lake NJ, DeSimone AM, Lek M. Neuromuscular disorders: finding the missing genetic diagnoses. Trends Genet. 2022;38(9):956–71.
Lee JD, Woodruff TM. The emerging role of complement in neuromuscular disorders. Semin Immunopathol. 2021;43(6):817–28.
Pratt J, De Vito G, Narici M, Boreham C. Neuromuscular junction aging: a role for biomarkers and exercise. J Gerontol A Biol Sci Med Sci. 2021;76(4):576–85.
Huijbers MG, Marx A, Plomp JJ, Le Panse R, Phillips WD. Advances in the understanding of disease mechanisms of autoimmune neuromuscular junction disorders. Lancet Neurol. 2022;21(2):163–75.
Clerc P, Lipnick S, Willett C. A look into the future of ALS research. Drug Discov Today. 2016;21(6):939–49.
Khamaysa M, Pradat PF. Status of ALS treatment insights into therapeutic challenges and dilemmas. J Pers Med. 2022. https://doi.org/10.3390/jpm12101601.
Smith AST, McCain ML, Bothwell M, Mack DL. Editorial: Modeling neuromuscular diseases to determine molecular drivers of pathology and for drug discovery. Front Cell Dev Biol. 2022;10:1017356.
Fralish Z, Lotz EM, Chavez T, Khodabukus A, Bursac N. neuromuscular development and disease: learning from in vitro and in vivo models. Front Cell Dev Biol. 2021;9: 764732.
Barbeau S, Tahraoui-Bories J, Legay C, Martinat C. Building neuromuscular junctions in vitro. Development. 2020. https://doi.org/10.1242/dev.193920.
Strickland JB, Davis-Anderson K, Micheva-Viteva S, Twary S, Iyer R, Harris JF, Solomon EA. Optimization of application-driven development of in vitro neuromuscular junction models. Tissue Eng Part B Rev. 2022;28(6):1180–91.
Lynch E, Peek E, Reilly M, FitzGibbons C, Robertson S, Suzuki M. Current progress in the creation, characterization, and application of human stem cell-derived in vitro neuromuscular junction models. Stem Cell Rev Rep. 2022;18(2):768–80.
Guo X, Gonzalez M, Stancescu M, Vandenburgh HH, Hickman JJ. Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials. 2011;32(36):9602–11.
Osaki T, Uzel SGM, Kamm RD. On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat Protoc. 2020;15(2):421–49.
Afshar Bakooshli M, Lippmann ES, Mulcahy B, Iyer N, Nguyen CT, Tung K, Stewart BA, van den Dorpel H, Fuehrmann T, Shoichet M, et al. A 3D culture model of innervated human skeletal muscle enables studies of the adult neuromuscular junction. elife. 2019. https://doi.org/10.7554/eLife.44530.
Faustino Martins JM, Fischer C, Urzi A, Vidal R, Kunz S, Ruffault PL, Kabuss L, Hube I, Gazzerro E, Birchmeier C, et al. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell. 2020;26(2):172–86.
Barik A, Li L, Sathyamurthy A, **ong WC, Mei L. schwann cells in neuromuscular junction formation and maintenance. J Neurosci. 2016;36(38):9770–81.
Beirowski B, Babetto E, Golden JP, Chen YJ, Yang K, Gross RW, Patti GJ, Milbrandt J. Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat Neurosci. 2014;17(10):1351–61.
Fuertes-Alvarez S, Izeta A. Terminal schwann cell aging: implications for age-associated neuromuscular dysfunction. Aging Dis. 2021;12(2):494–514.
Brosius Lutz A, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, Posada S, Zuchero JB, Barres BA. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci USA. 2017;114(38):E8072–80.
Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, Ward ME. Transcription factor-mediated differentiation of human iPSCs into neurons. Curr Protoc Cell Biol. 2018;79(1): e51.
Bettadapur A, Suh GC, Geisse NA, Wang ER, Hua C, Huber HA, Viscio AA, Kim JY, Strickland JB, McCain ML. Prolonged culture of aligned skeletal myotubes on micromolded gelatin hydrogels. Sci Rep. 2016;6:28855.
Denes LT, Riley LA, Mijares JR, Arboleda JD, McKee K, Esser KA, Wang ET. Culturing C2C12 myotubes on micromolded gelatin hydrogels accelerates myotube maturation. Skelet Muscle. 2019;9(1):17.
Lin YL, Nhieu J, Liu PY, Le G, Lee DJ, Wei CW, Lin YW, Oh SH, Lowe D, Wei LN. CRABP1-CaMKII-Agrn regulates the maintenance of neuromuscular junction in spinal motor neuron. Cell Death Differ. 2022;29(9):1744–56.
Chal J, Pourquie O. Making muscle: skeletal myogenesis in vivo and in vitro. Development. 2017;144(12):2104–22.
Thery M, Piel M. Adhesive micropatterns for cells: a microcontact printing protocol. Cold Spring Harb Protoc. 2009;2009(7):prot5255.
Acknowledgements
We like to acknowledge University of Minnesota Viral Vector Core (VVC), Physiology Core, University Imaging Center (UIC) and staff support from Dr. Guillermo Marques and Dr. Mary Brown, and the Minnesota Nano Center (MNC) supported by the National Science Foundation through the National Nanotechnology Coordinated Infrastructure (NNCI) under Award Number ECCS-2025124. Graphic abstract and figures were Created with BioRender.com. DJL and S-HO acknowledge support by the Minnesota Environment and Natural Resources Trust Fund as recommended by the Legislative-Citizen Commission on Minnesota Resources (LCCMR) and the Sanford P. Bordeau Chair in Electrical Engineering.
Funding
These studies were supported by NIH grants NS132277, DK54733, DK60521, and the Dean’s Commitment and the Distinguished McKnight Professorship of the University of Minnesota to LN.W. J.N. is supported by NIH fellowship F31DK123999.
Author information
Authors and Affiliations
Contributions
YLL designed and performed experiments and analyzed data. JN performed experiments and analyzed data; TL, LM, CWW performed experiments; DJL and SHO provided reagents, materials, and technical input. Stanley Thayer provided reagents and materials and technical input. LNW designed the experiments, analyzed the data, coordinated the study and provided financial support. LNW is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary data. Fig. S1.
The morphology of less differentiated MN1-derived MNs. Fig. S2. Image of 3D tri-culture on differentiation day 12. Fig S3. Relative positions of MNs and contracting myotubes for video 1.
Additional file 2: Video S1. Shows the MNs driving-myotube contraction under a bright field.
Additional file 3: Video S2. Shows an individual MU and the MN driving-myotube contraction under a bright field using a fluorescence filter for Texas Red.
Additional file 4: Video S3. Tri-culture with spontaneous and ACh induced contraction. Images show the 340 nm channel (left) and pseudo-color of the fura-2 340/380 fluorescence ratio (right). Increases in [Ca2+]i are indicated by red in the pseudo-color image. Initially, spontaneous contractions are visible at baseline. ACh (10 µM) application stimulated physical contraction (left) and an increase in [Ca2+]i.
Additional file 5: Video S4. Tri-culture with ACh induced contraction. Images show the 340 nm fluorescence (left) and pseudo-color of the fura-2 340/380 intensity ratio (right). Increases in [Ca2+]i are indicated by red in the pseudo-color image. ACh (10 µM) evoked physical contraction (left) and an increase in [Ca2+]i.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, YL., Nhieu, J., Lerdall, T. et al. A novel 3D bilayer hydrogel tri-culture system for studying functional motor units. Cell Biosci 13, 168 (2023). https://doi.org/10.1186/s13578-023-01115-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01115-2