
Abstract
Background
Autism Spectrum Disorders (ASD) patients experience disturbed nociception in the form of either hyposensitivity to pain or allodynia. A substantial amount of processing of somatosensory and nociceptive stimulus takes place in the dorsal spinal cord. However, many of these circuits are not very well understood in the context of nociceptive processing in ASD.
Methods
We have used a Shank2−/− mouse model, which displays a set of phenotypes reminiscent of ASD, and performed behavioural and microscopic analysis to investigate the role of dorsal horn circuitry in nociceptive processing of ASD.
Results
We determined that Shank2−/− mice display increased sensitivity to formalin pain and thermal preference, but a sensory specific mechanical allodynia. We demonstrate that high levels of Shank2 expression identifies a subpopulation of neurons in murine and human dorsal spinal cord, composed mainly by glycinergic interneurons and that loss of Shank2 causes the decrease in NMDAR in excitatory synapses on these inhibitory interneurons. In fact, in the subacute phase of the formalin test, glycinergic interneurons are strongly activated in wild type (WT) mice but not in Shank2−/− mice. Consequently, nociception projection neurons in laminae I are activated in larger numbers in Shank2−/− mice.
Limitations
Our investigation is limited to male mice, in agreement with the higher representation of ASD in males; therefore, caution should be applied to extrapolate the findings to females. Furthermore, ASD is characterized by extensive genetic diversity and therefore the findings related to Shank2 mutant mice may not necessarily apply to patients with different gene mutations. Since nociceptive phenotypes in ASD range between hyper- and hypo-sensitivity, diverse mutations may affect the circuit in opposite ways.
Conclusion
Our findings prove that Shank2 expression identifies a new subset of inhibitory interneurons involved in reducing the transmission of nociceptive stimuli and whose unchecked activation is associated with pain hypersensitivity. We provide evidence that dysfunction in spinal cord pain processing may contribute to the nociceptive phenotypes in ASD.
Similar content being viewed by others


Background
Autism Spectrum Disorders (ASD) are characterized by a range of sensory abnormalities, in particular in the domain of tactile sensitivity [1]. Among these, abnormal nociception is strikingly common in ASD and it manifests itself either as hypo-sensitivity or as hyper-sensitivity to painful stimuli. The common occurrence of self-injury, self-mutilation (including cases of self-extraction of teeth) and unreported wounds [2], supported by clinical and experimental studies [3,4,5] has been interpreted as evidence of reduced sensitivity to painful stimuli in ASD patients. Conversely, a subset of ASD patients display hyperalgesia and pain hypersensitivity in the form of mechanical/tactile allodynia [6], static mechanical allodynia (pain in response to light touch/pressure [7, 8], dynamic mechanical allodynia (pain in response to stroking lightly), reduced threshold for thermal pain [9], movement allodynia (pain triggered by normal movement of joints or muscles) and chronic pain unrelated to medical conditions [10,11,12]. Pain hypersensitivity may constitute a major and underappreciated source of discomfort for ASD patients, in particular due to their inefficient communication capabilities that afflict most of the affected ASD patients [2], resulting in unnecessary medical procedures [10].
It is worth noting that ASD mouse models, carrying mutations in different genes, may display hyper- or hyposensitivity to pain, suggesting that the variability of the clinical phenotype may be linked to the genetic heterogeneity of ASD (more than 800 genes are linked to ASD [13]). In fact, each genetic mutation might disrupt a discrete but different node of the nociceptive circuit, leading to mutation-specific phenotypes and mechanisms. In particular, loss of the ASD-related Shank3 protein results in pain hyposensitivity due to the disruption of the scaffold architecture, enabling TRPV1 signaling in dorsal root ganglion (DRG) neurons [14].
Since disturbed nociception is a major source of discomfort for patients, knowledge of the involved neuronal circuits may provide insights into diagnosis and treatment. Although substantial processing of somatosensory and nociceptive stimuli takes place in circuits in the dorsal spinal cord [15], we largely ignore the extent of the involvement of these circuits in ASD-associated nociceptive phenotypes, either in terms of the cellular subpopulations involved or in terms of molecular and neurochemical abnormalities at work. One could speculate that, since a large fraction of ASD-associated genes code for synaptic proteins [16] change in the synaptic architecture, connectivity and excitation/inhibition balance may be altered in the spinal cord of ASD patients and murine models.
Here we consider the ASD model obtained by deleting the gene coding for the postsynaptic density (PSD)-enriched scaffold protein Shank2 [17]. Indeed, point mutations and missense mutations in Shank2 are responsible for a small but consistent fraction of ASD cases [18].
Shank2−/− mice (obtained by targeting exon-6 and exon-7; [19, 20]), are considered a bona fide mouse model of ASD. In fact, they display a phenotype characterized by reduced social interaction, increased anxiety and compulsive grooming. Recent work has suggested that Shank2−/− mice may display abnormal nociception due to alteration of spinal cord circuits [21], although it must be stressed that deletion of different exons in distinct Shank2−/− models may also lead to different phenotypes [22]. Nevertheless, the neuronal subpopulations and the circuit architectural features responsible for such phenotype remain to be investigated.
Here we show that Shank2−/− mice display hypersensitivity to formalin-induced pain; notably, we have identified (in murine and human spinal cord) a subpopulation of glycinergic interneurons characterized by very high expression levels of Shank2. Loss of Shank2 results in the reduction in synaptic NMDAR and in the blunted recruitment of these inhibitory interneurons upon painful stimuli, which results in the over-activation of lamina-I nociceptive projection neurons.
Materials and methods
Animals
All experiments were performed in agreement with the local and national guidelines for animal experimentation. In particular, all experiments were approved by the Regierungspräsidium Tübingen under the licence n° o.103 and by the Italian Ministry of Health 966/2016-PR.
All transgenic mouse lines used were previously described. Glycine transporter 2 (GlyT2)-EGFP mice [23] tissue samples were a courtesy of Silvia Arber; spinal cord samples from parvalbumin (PV)-Cre x tdTomato-ROSA26 [24] were a courtesy of Pico Caroni. Vesicular GABA transporter (VGAT)-Cre x tdTomato-ROSA26 [25], pancreas associated transcription factor 1a (Ptf1a)-Cre x tdTomato-ROSA26 [26], paired related homeobox protein-like 1 (Prrxl1)-Cre x tdTomato-ROSA26 (also known as DRG11); [27] and vesicular glutamate transporter 2 (vGluT2)-CRE x tdTomato-ROSA26 [25] double transgenic mice were a courtesy of Filippo Rijli, Ahmad Bechara and Alberto Loche. Ret-GFP transgenic mice were previously reported [28]. Shank2−/−(Δ7) mice were previously described [19].
Shank2 antibodies
Polyclonal rabbit antiserum against the C-terminus of Shank2 (SA5192), custom made, was previously described [29]. Briefly, partial cDNAs of the ProSAP1 cDNA (encoding amino acids 826–1259) were subcloned into the bacterial expression vector pGEX-1T (Pharmacia Biotech, Uppsala, Sweden). A 95 kDa glutathioneS-transferase (GST)-ProSAP1 fusion protein was expressed in Escherichia coli XL 1 Blue and used to immunize rabbits.
pEGFP–ProSAP1 construct
pEGFP–ProSAP1 constructs were provided by Prof Boeckers and was previously reported [30]. Full-length ProSAP1 cDNAs were cloned into the pEGFP (C1-3) vector (Clontech, Palo Alto, CA, USA) coding for fusion proteins with the GFP at the N-terminus.
HEK cell culture and Shank2 transfection and overexpression
HEK cells (Human embryonic kidney 293 cells) were cultured in 6-well plates in DMEM (10% FCS and 1% Pen/strep) with a density of 80.000 cells per well. 1 day after plating, cells were transfected with 1 µg of shank2 plasmid (pEGFP-ProSAP1) with PEI protocol [31] overnight, followed by change of medium, washing and harvesting of cells. Extraction of protein was performed as described below [32, 33].
Western blot
Western blot for Shank2 was performed as previously reported [32]. Briefly, the lumbar spinal cord, cortex, cerebellum, hippocampus, striatum, and dorsal root ganglia were dissected from wild type (WT) and Shank2−/− mice (sacrificed by cervical dislocation). Tissue samples were snap-frozen on dry ice and quickly homogenized in complete radioimmunoprecipitation assay (RIPA) buffer with Protease and Phosphatase inhibitors using a mortar. The homogenate was subject to sonication and then cleared by centrifugation (10,000 g, 10 min, 4 °C). Protein concentration was measured by BCA kit and 20 µg of protein were loaded in each lane of an 8%-acrylamide gel. As a positive control, protein extract (2 µL) from an overexpression of Shank2, using a plasmid (pEGFP-ProSAP1), in HEK cells was used. As negative control proteins from untransfected HEK cells were used. Proteins were transferred to a nitrocellulose membrane using a Trans-Blot® Turbo™ Transfer System (Bio-Rad) semi-dry transfer apparatus (standard protocol for mixed molecular weight was run twice); membranes were blocked in 5% bovine serum albumin (BSA) in tris buffered saline (TBS) added with 1% Tween20 for 1 h and incubated with anti-Shank2 SA5192 antibody (diluted 1:500 in blocking solution) overnight at 4 °C on an orbital shaker. After 6 washing steps of 20 min with TBS added with 0.25% Tween the membranes were incubated with a horseradish peroxidase (HRP) coupled secondary antibody from Bio-Rad® for 1 h at room temperature, diluted in blocking solution (5% BSA in TBS added with 1% Tween20). The HRP coupled blots were then incubated in Clarity Western enhanced chemiluminescent (ECL) HRP substrate solution (Bio-Rad) for 5 min. Detection was performed with a digital CCD-camera based system from Fuji (Tokio, Japan). Membranes were then re-incubated at room temperature for 1 h with anti-ꞵ-tubulin antibody (1:1000 in blocking buffer). Quantification was performed with imageJ Fiji toolbox, normalizing each band on ꞵ-tubulin.
Intraspinal injection of AAV9-GFP for sparse labelling
Intraspinal injection of adeno associated virus (AAV9) was performed as previously reported [34]. Briefly, WT mice aged P20 were administered buprenorphine (0.05 mg/Kg) and meloxicam (0.1 mg/Kg) 30 min before being anesthetized with sevoflurane (4% in 96% O2, 800 ml/min). Dorsal skin was shaved and incised (10 mm) at lumbar level. Paraspinal muscles were blunt-dissected and dorsal laminae were sectioned at vertebrae T11-T13 level. Upon removal of the dorsal laminae bone flap, the underlying dura was opened using a 33G needle and washed with artificial cerebrospinal fluid (ACSF). Viral injections were performed at the coordinates (y = + 0.22–25 mm; z = − 0.55 to 6 mm) having the central dorsal artery as reference. A total of four injections were performed, 250 nl/5 min each, using a pulled-glass capillary connected to a Picospritzer-III apparatus. Muscles were thereafter sutured on the midline using Prolene 7.0 surgical thread; the fascia was repositioned to cover the wound, and the skin was stiched on the midline using Prolene 6.0 surgical thread. Mice were then transferred to a warmed recovery cage with facilitated access to food and water and were administered additional doses of buprenorphine every 12 h for the following 72 h.
CTB retrograde labeling
Retrograde labeling of sensory neurons and their central afferents in the dorsal spinal horn was performed as previously reported [35]. Briefly, adult WT mice were anesthetized using isoflurane, and the injection site was shaved. 0.1–0.3 µl of Choleratoxin subunit B (CTB) conjugated with Alexa Fluor 488 (2 µg/µl in phosphate buffered saline (PBS); Thermofischer) was injected into the hairy skin of leg using a fine glass capillary. Mice were sacrificed 7 days after the injection and further processed.
Immunostaining
Spinal cord samples were processed as previously reported [36]. Briefly, mice were perfused with 4% paraformaldehyde (PFA) in PBS, (L1–L5) lumbar spinal cord was isolated and post-fixed for 18 h in 4% PFA. Tissue samples were thereafter washed and cryoprotected overnight at 4 °C in 30% sucrose. After embedding in optimal cutting temperature (OCT, Tissue-Tek), 40 µm cryostat sections were obtained. Subsequently sections were blocked, incubated for 48-72 h with primary antibodies (see Additional file 1: Table S1) diluted in PBS with 3% BSA, 0.3% Triton X-100 at 4 °C on an orbital shaker. Sections were then washed with PBS (30 × 3 min at RT) and incubated for 2 h at room temperature (RT) with appropriate combinations of secondary antibodies (see Additional file 1: Table S1) and DAPI (1:1000) for nuclear staining. For Isolectin GS-IB4 staining, a directly Alexa Fluor 488-conjugated antibody (Invitrogen, cat. 121411) was used to label nonpeptidergic nociceptive primary afferents in laminae II, at the concentration of 1:1000 diluted in 10% normal donkey serum (NDS) additioned with 0.3% Triton-x. After additional washing in PBS (3 × 30 min at RT), the sections were dried and mounted with Prolong antifade mounting medium (Invitrogen). For stimulated emission depletion (STED) Microscopy, before imaging, the samples were washed shortly with dH20 and mounted in 2,2-thiodiethanol (TDE) buffer.
Clarity and immunostaining on human spinal cord samples
Human spinal cord samples were obtained in agreement with the procedures approved by the ethical committee of Ulm University (n.245/17), full data on human samples is available in Additional file 1: Table S2. Clearing and immunostaining were performed on 100 µm thick post-mortem human lumbar spinal cord sections as previously described [37]. Briefly, sections were incubated in hydrogel solution (40% acrylamide, 4% PFA, 0.25% VA-044 initiator; pH = 7.3) at 4 °C for 1 week. Sections were then degassed using a desiccator and polymerized at 37 °C for 1.5 h until the hydrogel has completely hardened. The excess of hydrogel was afterward removed by washing for 1.5 h at 37 °C with PBS. The samples were then passively cleared in a clearing solution (4% SDS, 200 mM boric acid solution; pH = 7.3) for 1 week at 37 °C. The clearing solution was afterward washed away for 1 day at 37 °C with 0.1% TritonX-100. Once cleared, the sections were stained starting with a blocking step in 3% BSA and 0.3% TritonX-100 for 2 h at RT. After blocking, sections were incubated in primary antibody (Additional file 1: Table S1) for 72 h at 4 °C followed by a washing step in 0.1% TritonX-100 3 times for 30 min. Subsequently, sections were incubated in secondary antibodies for 2 h at RT, and washed 3 times for 30 min in 0.1% TritonX-100. Sections were finally mounted on microscope glass slides with Prolong antifade mounting medium (Invitrogen).
Overview images of the spinal cord sections were taken with a fluorescent microscope (Keyence, BZ-X800) equipped with a 4 × air objective. Higher resolution images were then performed using a laser-scanning confocal microscope (Zeiss LSM 980, Carl Zeiss), with a 20 × air or 100 × oil objective, by taking tile-scan images of the grey matter or single neurons in laminae I–IV.
In situ single-mRNA hybridization
Fluorescence in situ single mRNA hybridization [38] was performed according to manufacturer instructions (Fluorescence In Situ mRNA Hybridisation for fixed frozen tissue, RNAscope by ACDBio) with small modifications (as previously reported; [39]). Shortly, sections were mounted and dried on glass slides, quenching of autofluorescence was performed by pretreatment with 0.1 M Glycine in PBS for 15 min. Thereafter, a 3 min antigen retrieval step was performed, and sections were washed twice in dH2O and once in ethanol. Then pretreatment reagent III (all reagents were provided by ACDbio) was applied for 20 min at 40 °C. The Probes (GlyT2 and c-FOS) were hybridized for 4.5 h at 40 °C, followed by a wash step of 2 × 2 min at RT. The sections were then incubated for 30 min with amplification-1 reagent at 40 °C, followed by a wash step of 2 × 2 min at RT. Subsequently, the sections were incubated for 15 min with amplification-II reagent at 40 °C, and washed 2 × 2 min at RT. The last amplification step was performed by incubating the sections for 30 min with amplification-III reagent at 40 °C, followed by a wash step of 2 × 2 min at RT. After this, the sections were incubated with amplification-IV for 45 min at 40 °C, followed by a final wash step of 2 × 10 min at RT. Sections were counterstained with DAPI or co-immunostained with specific markers of interest. For the co-immunostaining, the sections were blocked (3% BSA, 0.3% Triton in PBS) for 1 h, then incubated overnight at 4 °C with primary antibodies, diluted in blocking buffer (see Additional file 1: Table S1). After the incubation the sections were washed for 3 × 30 min in 1X PBS. Subsequently, a 2 h incubation with secondary antibody diluted in the blocking buffer at RT (Donkey anti-guinea pig 568, 1:500 [invitrogen]) was performed. After the last washing steps (3 × 30 min in PBS), the sections were counterstained with DAPI and mounted using Fluorogold prolong antifade mounting medium (Invitrogen).
Stimulated emission depletion microscopy (STED)
A custom build dual-color setup, with a high NA objective (HCX PL APO 100x/1.40–0.70 oil CS, Leica), was used for STED microscopy as described previously [40]. Briefly, a supercontinuum laser source (SC-450-PP-HE, Fianium) with a broad spectral range provided all the excitation (568 nm and 633 nm) and depletion (~ 720 nm and ~ 750 nm) beams, samples were scanned with a piezo stage (733.2DD, Physik Instrumente) and emission was recorded by an avalanche photodiode (SPCMAQRH-13/14-FC, Perkin- Elmer). Acquisition mode could be switched between confocal (diffraction limited resolution) or STED (lateral resolution ~ 35 nm) and was controlled by a custom LabVIEW (National Instruments) software.
Confocal imaging
Confocal images were acquired using a LSM-710 or LSM-980 (Carl Zeiss AG) microscope as previously reported [34], fitted with a 20× air objective or with 40×, 60× or 100× oil objectives with optical thickness fitted to the optimum value. For overview images, 20× objective 6 × 6 image tiles were acquired. For high-magnification images, a zoom factor 3× was applied during the acquisition of 60× or 100× oil objective images, oversampled in the z-axis to twice the theoretical optimal value. All images have been acquired at 1024 × 1024 pixels resolution and 12- or 16-bit depth. Acquisition parameters were set to avoid over or under-saturation and kept constant for each experimental set.
Image analysis
For image analysis, confocal pictures were loaded into ImageJ. For the definition of Shank2 immunostaining intensity, a circular region of interest was manually located around each neuronal nucleus (identified in NeuN staining) and the integrated fluorescence intensity (expressed in arbitrary units -au- ranging between 0 and 4095) averaged over the area of the region of interest, was logged. For the identification of Shank2high neurons, we defined a threshold by considering the top of the distribution of intensities in laminae I and II and adding 200 au, thus defining as Shank2high any neuron whose average fluorescence was at least 200 au higher than the brightest neuron in laminae I and II. Operationally, Shank2 immunofluorescence images were thresholded until neurons in laminae I and II were no longer visible and the threshold value was then moved 200 au higher; any Shank2 + neuron still visible at this stage was considered Shank2high. In the quantification of Shank2 expression in human samples, the contour of neurons was manually drawn using the SMI-311 immunostaining as reference and the average Shank2 immunostaining intensity was quantified. For the analysis of Homer, GluN1, vGluT1 and vGluT2 cluster size on GlyT2 + interneurons, the contour of each interneuron was manually drawn in imageJ; after thresholding, all clusters juxtaposed to the contour were highlighted and the size logged. The analysis of the c-Fos mRNA was performed by counting the single c-Fos mRNA dots per GlyT2 + interneuron (depicted with GlyT2 mRNA). Since c-Fos was expressed in every GlyT2 + neuron, we defined a threshold of at least 50 mRNA molecules per cell for it to be considered c-Fos positive. The neuronal and synaptic architecture was analysed by taking a region of interest in the predefined laminae and counting manually the number of neurons and with the Imaris software the density of synapses in the region of interest.
Formalin test
Formalin test was performed according to the previously reported protocols [41, 5d, e). Finally, immunostaining on postmortem human spinal cord revealed that the post-synaptic Shank2 terminals receive pre-synaptic input of vGluT1, 36.9 ± 14.1% of Shank2 synapses were positive for vGlut1 (Fig. 5f), which was comparable to the mouse data.
Thus, Shank2high receive direct somatosensory input but Shank2 + synapses do not correspond to a distinct type of input.
Neuronal architecture of the spinal cord is unaffected by Shank2−/−
Having established the existence and the nature of a new Shank2high subpopulation of glycinergic interneurons, we set out to investigate how the loss of Shank2 in Shank2−/− mice may affect these interneurons and generate the pain-hypersensitivity phenotype observed in behavioural tests. Since several transgenic mouse lines with sensory or nociceptive phenotypes are characterized by disruption or selective loss of neuronal subpopulations [41, 62] we verified that loss of Shank2 did not result in any gross disturbance of spinal cord architecture. In fact, Two-way ANOVA (Bonferroni corrected) revealed that the overall density of neurons (NeuN+; F(1.16) = 3.977; p > 0.05; Additional file 1: Fig. S5a, b) and in particular of inhibitory neurons (Pax2+; F(1.16) = 0.3959; p > 0.05) in the dorsal laminae of the spinal cord was comparable in WT and Shank2−/− (Additional file 1: Fig. 5c, d). Furthermore, unpaired t-test revealed that the density of excitatory interneurons of laminae II (Protein kinase C gamma+ (PKC-γ+); t = 2.296; df = 4; p > 0.05) was comparable in WT and Shank2−/− (Additional file 1: Fig. 5e, f). Likewise, the number of neurons expressing GlyT2 mRNA was similar in WT and Shank2−/− mice (F(1.16) = 0.2588; p > 0.05; Additional file 1: Fig. 5g, h).
Finally, we determined the density of inhibitory synapses across the dorsal laminae; no significant difference has been found between WT and Shank2−/− in the number of synapses per area (synaptic density) for GlyT2+ puncta (Two-way ANOVA: F(1.30) = 0.8204; p > 0.05; Additional file 1: Fig. S6a, b), VGAT+ puncta (F(1.20) = 0.004215; p > 0.05; Additional file 1: Fig. S6c, d) and gephyrin+ puncta (F(1.48) = 3.626; p > 0.05; Additional file 1: Fig. S6e, f). Thus, deletion of Shank2 did not cause any obvious changes in the overall architecture of the dorsal horn.
Loss of Shank2 disrupts NMDAR clustering in excitatory synapses on glycinergic interneurons
Since Shank2 is a core scaffold protein in glutamatergic synapses, we reasoned that excitatory synapses on Shank2high glycinergic interneurons may suffer substantial postsynaptic (and/or presynaptic) alterations in Shank2−/− mice. We identified glycinergic interneurons by the detection of GlyT2 in situ hybridization and co-immunostained the sections for postsynaptic proteins and receptors. This approach provides a representative value of the synaptic inputs, within the limitation imposed by the consideration of the cell body only (dendrites are not easily visible with this approach). Because of the protease treatment required for mRNA detection, only a restricted number of antibody-antigen couples (whose antigens were protease-resistant) could be employed. In particular, we considered the GluN1 NMDAR subunit and the PSD scaffold protein Homer. Interestingly, the number of GluN1 and Homer post-synaptic structures on GlyT2 interneurons was comparable in WT vs Shank2−/− mice (95.3 ± 17.5 vs. 107.4 ± 31 per 100 µm WT vs. Shank2−/−; unpaired t-test; t = 1.034; df = 6; p > 0.05 for GluN1; 80.1 ± 20.2 vs. 104.7 ± 25.8 per 100 µm WT vs. Shank2−/−; unpaired t-test; t = 1.010; df = 6; p > 0.05; for Homer) indicating that the number of synapses and their qualitative composition were not affected by Shank2 loss. However, GluN1 clusters were significantly smaller in glycinergic interneurons of Shank2−/− mice compared to WT mice (WT vs. Shank2−/−; 0.36 ± 0.06 µm2 vs. 0.27 ± 0.01 µm2; unpaired t-test; t = 3.229; df = 6; p < 0.05; Fig. 5g, h). Remarkably, this reduction in size was not observed for Homer (WT vs. Shank2−/−; 0.36 ± 0.36 µm2 vs. 0.32 ± 0.32 µm2; unpaired t-test; t = 0.5888; df = 4; p > 0.05; Fig. 5i, j).
Since Shank2 proteins may regulate trans-synaptic signaling, we also took into consideration the number and the size of presynaptic excitatory boutons. To this aim, glycinergic interneurons were identified by GlyT2 in situ hybridization and vGlut1+ and vGlut2 + terminals by immunostaining. The number of vGlut1 and vGlut2 terminals on GlyT2 interneurons was comparable in WT and Shank2−/− mice (16.3 ± 5.2 vs. 17.8 ± 7.8 per 100 µm WT vs. Shank2−/−; unpaired t-test; t = 0.3373; df = 6; p > 0.05 for vGlut1; 59.2 ± 17.7 vs. 64.2 ± 18.8 per 100 µm WT vs. Shank2−/−; unpaired t-test; df = 6; p > 0.05 for vGlut2); however, vGluT2 terminals were significantly smaller in Shank2−/− mice compared to WT mice (WT vs. Shank2−/−; 0.47 ± 0.06 µm2 vs. 0.36 ± 0.06 µm2; unpaired t-test; t = 2.599; df = 6; p < 0.05; Fig. 5m, n). This difference was not observed in the vGluT1 terminals (WT vs. Shank2−/−; 0.86 ± 0.69 µm2 vs. 0.79 ± 0.78 µm2; unpaired t-test; t = 0.9864; df = 6; p > 0.05; Fig. 5k, l).
Taken together these data show that loss of Shank2 does not affect the number of excitatory synapses on glycinergic interneurons but causes the selective decrease of synaptic NMDAR content and in the size of propriospinal glutamatergic presynaptic terminals. Thus, Shank2 loss appears to selectively weaken glutamatergic input and plasticity on glycinergic interneurons.
Reduced activation of glycinergic interneurons in Shank2−/− mice is associated with increased excitation of dorsal laminae interneurons upon nociceptive stimulation
Next, we investigated if the disturbance in the structure of synaptic inputs to Shank2high interneurons affected their activation during nociceptive stimulation. To this aim, WT and Shank2−/− mice were injected with Formalin (or with saline) in the plantar aspect of the right hindpaw (as in the Formalin test) and sacrificed 120 min later (90 min after the onset of the phase-II). We used single-mRNA molecule in situ detection to simultaneously identify glycinergic interneurons (using GlyT2 mRNA) and c-fos mRNA expression as a marker of neuronal activity (the use of c-fos immunostaining was precluded by the protease treatment necessary for the identification of GlyT2 mRNA). Two-way ANOVA revealed a significant effect in the treatment groups (F(3,9) = 21.67; p = 0.0002). Post-hoc analysis (Bonferroni corrected) revealed that indeed, the number of c-fos + /GlyT2 + neurons was very low at baseline and comparable in WT and Shank2−/− samples (WT vs. Shank2−/−; 10.7 ± 4.7% vs. 9.8 ± 5.2%; Two-Way ANOVA; df = 9; p > 0.05, Fig. 6a, b); however, upon Formalin challenge, the number of double-positive interneurons increased strongly in WT animals but much less so in Shank2−/− (WT vs Shank2−/−; 50.6 ± 8.3% vs. 18.1 ± 8.9%; Two-Way ANOVA; df = 9; p < 0.01; Fig. 6a, b) implying a failure in GlyT2+ neuron activation upon nociceptive stimulation in the Shank2−/− mice.

Reduced activity in glycinergic interneurons is associated with increased activity of interneurons in the dorsal laminae I and II. Activation patterns in glycinergic interneurons and interneurons in dorsal laminae upon activation. Mice were treated with low concentration of Formalin and c-fos levels were measured 120 min later (a, b) Saline injection resulted in no difference in c-fos levels in glycinergic cells between WT and Shank2−/− mice (p > 0.05). Upon Formalin injection c-fos was significantly higher in WT vs Shank2−/− mice in glycinergic cells (p < 0.01); (N = 4) scale bar: 20 µm. c and d The reduced activity of glycinergic cells in Shank2−/− mice in turn significantly increased the overall c-fos expression in the Shank2.−/− in laminae I (p < 0.001), Laminae II (p < 0.01) but no difference in laminae III and IV–V (p > 0.05; p > 0.05) upon formalin injection; (N = 4) scale bar: 100 µm. Data shown as average ± SD. **p < 0.01; ***p < 0.001
We then sought to verify if the observed decrease in inhibitory interneurons activity would result in the overall increase in excitation in the dorsal-horn circuitry of Shank2−/− mice. We considered spinal cord sections obtained 120 min after a Formalin challenge in the right hindpaw; in order to monitor the large-scale activation of neurons in the dorsal spinal cord, the samples immunostained for NeuN (to identify all neurons) and c-Fos and the ratio of c-Fos+/NeuN+ cells was annotated according to the spinal cord lamina. Two-way ANOVA revealed a significant effect in treatment groups (F(1,48) = 62.82; p < 0.0001). Post-hoc analysis (Bonferroni corrected) revealed that, in saline-injected WT mice, c-Fos+ neurons were detectable in very low numbers across dorsal spinal cord (2.2 ± 1.1% in laminae I) and in saline-injected Shank2−/− mice the percentage of c-fos+ neurons were comparable to WT mice (4.5 ± 2.7% of NeuN+ in laminae I; p > 0.05). Formalin injection caused a much larger elevation of c-Fos+ neurons in Shank2−/− vs WT in laminae I (WT vs. Shank2−/−; 35.1 ± 4.5% vs. 75.1 ± 8.2%; Two-Way ANOVA; df = 24; p < 0.0001; Fig. 6c, d) as well as in laminae II (WT vs. Shank2−/−; 11.9 ± 5.0% vs. 24.6 ± 4.2%; Two-Way ANOVA; df = 24; p < 0.001; Fig. 6c, d); but no difference was found in laminae III, IV and V (WT vs. Shank2−/−; Laminae III: 16.9 ± 9.0% vs. 16.9 ± 3.6%; WT vs. Shank2−/−; p > 0.05; Laminae IV-V: 24.7 ± 5.8% vs. 23.0 ± 3.9%; Two-Way ANOVA; df = 24; p > 0.05; Fig. 6c, d). A comparable result was obtained when the number of cells positive for c-Fos mRNA (by in situ hybridization) was measured in the same groups (Two-way ANOVA: F(1.24) = 35.60; p < 0.0001); post-hoc analysis (Bonferroni) showed a significant difference between Shank2−/− versus WT in laminae I (WT vs. Shank2−/−; 4.0 ± 0.9 vs. 8.7 ± 1.4; per 104 µm2; p < 0.0001; Additional file 1: Fig. S7a, b), underscoring the robustness of the dataset.
Taken together, these data suggest that the disruption of the excitation of inhibitory interneurons in Shank2−/− mice results in a decreased excitation of glycinergic inhibitory interneurons upon acute chemical pain and, in turn, to the increased activation of neurons in the dorsal horn following a nociceptive stimulus.
Discussion
Our data show that high levels of Shank2 expression identifies a subpopulation of glycinergic inhibitory interneurons located in the dorsal horn of the spinal cord, receiving inputs from somatosensory afferents; Shank2 loss leads to a decrease in excitatory synapses onto glycinergic interneurons and in net increase of excitation in the dorsal horn, correlated with increased sensitivity to chemically induced pain.
The proposed mechanism involved in pain hypersensitivity of Shank2−/− mice is therefore related to dis-inhibition of spinal cord nociceptive circuits [63, 64]. A similar increase in nociception has been observed in several experimental models in which the inhibitory tone has been decreased: pharmacological blockade of glycinergic inhibition produces mechanical allodynia through the dis-inhibition of PKC-γ+ neurons [65] and silencing of glycinergic interneurons is sufficient to induce allodynia [59]. This concept has been further supported by recent data obtained in patients with loss-of-function mutations of glycine receptors or glycine transporters (leading to the clinical syndrome of hyperekplexia), who display decreased pain thresholds and amplified pain withdrawal reflexes [66]. Increased nociception as a consequence of loss of spinal inhibition has been also observed in case of loss of GABAergic inputs [67]: inactivation of PV+ inhibitory interneurons cause the appearance of tactile allodynia [68, 69], whereas GABAergic agonists produce anesthesia [70]. Within the conceptual framework of the “gate-control” theory, inhibitory inputs are well understood to prevent the runaway activation of PKC-γ+ interneurons by touch-evoked inputs as well as other excitatory interneurons in laminae II [15], which, in turn, would drive the excitation of laminae I (NK1+) interneurons. In agreement with this model, reduced excitation of glycinergic interneurons in Shank2−/− mice leads to increased activity of laminae I neurons.
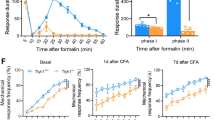
Abnormalities in pain processing have been previously reported in Shank2−/− mice [21, 71]. Ko et al. reported of an overall decreased basal tactile perception and acute pain response in Shank2−/− and induction of neuropathic or inflammatory chronic pain. Some of the reported findings agree with our own: the reduced mechanical allodynia at baseline reported by Ko is also visible in our dataset (Fig. 1d-pre) with a response comparable in the two genotypes up to day 7 in the CFA-von Frey test followed by reduced chronic pain at a later timepoint (Fig. 1d). Furthermore, Shank2−/− did display a longer latency to escape in the texture preference test (Fig. 1n), implying a decrease in tactile sensitivity. In other cases, conditions are not fully comparable: the hot-plate test was performed by Ko at 55 °C, whereas 45 °C was used in this study, and we did not specifically investigate neuropathic pain, which may hinge on different long-term sensitization mechanisms [72]. Taken together, ours and previously published data suggest that nociceptive abnormalities in Shank2−/− may be specific of sensory modalities and depending on multiple alterations in synaptic plasticity and circuit function.
Genetic approaches have increasingly revealed the high degree of heterogeneity in neuronal subpopulations in the dorsal spinal cord [41, 61, 62]. Recently, the genetic diversity of these populations has been demonstrated to be extensive and a large number of subpopulations have been identified in a single-cell transcriptome study [73]. Nevertheless, neuronal physiology is highly influenced by the quantity and quality of their synaptic inputs, which is strongly dependent upon the composition and architectural organization of postsynaptic structures [74, 75]. Therefore, a distinct layer of diversity may be identified once the synaptic composition of neuronal subtypes is taken into consideration. Here we provide a first proof of this concept; in fact, Shank2 distinguishes a subclass of glycinergic and parvalbumin interneurons as well as a small population of excitatory interneurons.
Since different members of the Shank family, despite their similarity, are not considered mutually redundant [19, 76], the function of Shank2high cells is predicted to be heavily impacted by Shank2 loss, despite their anatomical integrity (i.e., their normal number and positioning). Of note, the Shank2 gene gives rise to several splice variants and isoforms generated using alternative promoters (ranging from 130 to 230 KDa; [29, 53, 77]). The Shank2−/− model used in the present study is more precisely a deletion mutant lacking the exon 7 and this mutation should lead to the early termination and non-sense decay of the Shank2 mRNA [19]. In agreement with the cortex finding, in the spinal cord homogenate of Shank2−/− mice we observe the loss of the most abundant isoforms (160–220 kDa) whereas only a low abundance, low MW isoform may still be expressed. However, the immunostaining of cortical samples from Shank2−/− mice reveals a largely complete loss of immunoreactivity and the immunostaining of spinal cord from Shank2−/− mice reveals an almost complete loss of immunoreactivity (any residual immunoreactivity seen in spinal cord may correspond to the low-abundance 130 KDa isoform, whose functional relevance, if any, remains to be investigated).
DRG are known to express some isoforms of Shank3, which contribute to arrange the arrays of receptors and ion channels in the peripheral projections of the ganglion cells [14, 54]. However, DRGs appear to express very little of the 160–180 KDa better known Shank2 isoform, and almost none of the 160 kDa isoform which is characteristically eliminated by the deletion of the exon 7. The immunostaining of DRG for Shank2 revealed only a small number of cells with a very faint immunopositivity, in contrast with the abundant and widespread expression of Shank3 [14]. While it is not possible to fully exclude any DRG or peripheral effect of Shank2 loss on sensory phenotypes observed in our mouse model, the limited expression of Shank2 in DRG suggest that at least a component, possibly a substantial one, of the observed phenotype is due to the loss of Shank2 within the central nervous system (CNS).
In agreement with observations in other neuronal subtypes [22, 76, 78], we find that loss of Shank2 causes the decrease in NMDAR expression in excitatory synapses on GlyT2+ interneurons. Although baseline neurotransmission in the pain processing circuit appears to be not affected (as the acute phase of the formalin test is comparable in Shank2−/− mice and WT littermates), the NMDAR-dependent synaptic plasticity that is thought to underlie the second phase of the behavioural response to the formalin test [79,80,81] may be unbalanced, with insufficient potentiation of the inhibitory circuit. In fact, glycinergic interneurons are strongly activated in WT (as shown by the c-Fos induction), but not in the Shank2−/− animals. Thus, the resulting decrease in glycinergic transmission in the pain processing circuit would cause excessive excitatory drive, as demonstrated by the increase in the number of c-fos+ neurons in laminae I and II. In fact, dysfunction or silencing of inhibitory interneurons is known to cause pain hypersensitivity in human patients and experimental animals [59, 66, 82] in particular by dis-inhibiting PKC-γ+ excitatory interneurons [65]. However, disturbances in excitatory synapses due to Shank2 loss may affect other neuronal elements of the circuit: in fact, pain induced by intrathecal administration of NDMA is reduced in Shank2−/− mice, suggesting that Shank2 loss may affect differentially multiple sensory and nociceptive modalities.
Thus, circuits involved in nociception may be disturbed across multiple nociceptive stimuli and alteration of somatosensation may co-exist (as previously shown, [54, 83]). Interestingly, Shank2−/− mice have been also reported to display a reduced sensitivity to the nociceptive response evoked by intrathecal injection of NMDA [21]. Since this procedure does not selectively activate one subpopulation of neurons, it is not straightforward to explain it in circuit terms. In fact, Shank2 is highly enriched in glycinergic interneurons, but it is not restricted to these cells (see Fig. 1a, b and Additional file 1: Fig. S1) and even among the Shank2high cells, a fraction of excitatory neurons (VGluT2+, Prrxl1+) can be identified and their role remains unexplored. Furthermore, the dysfunction of PV interneurons in spinal cord has been directly related to mechanical but not thermal allodynia[69]; interestingly, only a fraction of PV interneurons appear to be Shank2high and indeed we detect thermal but not mechanical allodynia.
Thus, the impact of Shank2 loss may affect modality-specific pain processing circuits in a distinct way, depending on the role of different cellular subpopulations.
The insufficient activation of glycinergic interneurons because of disrupted excitatory synapses observed in Shank2−/− mice is a new mechanism for abnormal pain processing in autism. In fact, reported pain hyposensitivity in Shank3 mice has been linked to the loss of Shank3 in neurons in the dorsal root ganglia and the direct effect of Shank3 absence on TRPV channel expression [14]. Likewise, autism-related behavioural dysfunctions have been linked to the disturbed sensory input generated by abnormal sensory neurons in dorsal root ganglia [54, 83]. Conversely, loss of function of the ASD-associated gene Caspr2 is associated with neuropathic pain [84] through mechanisms involving increased sensitization of neurons in dorsal root ganglia. Although these reports all point to a sensory dysfunction originating in the periphery, our findings suggest that disruption of spinal cord circuits may be a strong contributor to the observed hyper- and hyposensitivity to nociceptive stimuli. Furthermore, one can speculate that the same excitation/inhibition balance disruption that is thought to underlie the ASD spectrum disorder may also manifest itself in spinal circuits to contribute to drive sensory abnormalities.
Limitations
The findings of this study must be considered within the boundaries of a few limitations. First, we explored the nociceptive phenotype only in male mice; since ASD is more prevalent in males (3:1, [85]), our findings are relevant to the majority, but not necessarily to all, of ASD patients. Nevertheless, gender dimorphism in pain sensitivity has been reported [86, 87] and extrapolation of our results to females has to be cautious. Second, ASD patients display a large genetic heterogeneity [13] and a very diverse phenotype in nociception, ranging from hyposensitivity to hypersensitivity. Thus, our findings may not necessarily apply to ASD related to mutations in genes other than Shank2.
Conclusion
In conclusion, we demonstrate that the Shank2 expression level characterizes a subset of inhibitory interneurons whose activation is disrupted by Shank2 loss leading to excessive nociceptive circuit activation and allodynia. Although Shank3 expression is known to be compensatory increased upon Shank2−/− [19], no functional compensation takes places in glycinergic interneurons, whose excitatory synapses appear to be functionally and structurally disrupted. It remains unclear which role of Shank2 is absolutely required in these cells that cannot be compensated by other Shank family members. Our findings also suggest that treatment of nociceptive disturbances in ASD may require to be tailored to the underlying genetic cause, even when phenotypes converge, since distinctive pathophysiology may be at play in each case.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- AAV:
-
Adeno associated virus
- ACSF:
-
Artificial cerebrospinal fluid
- ASD:
-
Autism spectrum disorder
- BSA:
-
Bovine serum albumin
- CFA:
-
Complete Freund adjuvant
- CNS:
-
Central nervous system
- CTB:
-
Cholera toxin subunit B
- DRG:
-
Dorsal root ganglion
- ECL:
-
Enhanced chemiluminescent
- GlyT2:
-
Glycinergic transporter 2
- HRP:
-
Horseradish peroxidase
- NDS:
-
Normal donkey serum
- OCT:
-
Optimal cutting temperature
- PBS:
-
Phosphate buffered saline
- PFA:
-
Paraformaldehyde
- PKC-y:
-
Protein kinase C gamma
- Prrxl1:
-
Paired related homeobox protein-like 1
- PSD:
-
Postsynaptic density
- Ptf1a:
-
Pancreas associated transcription factor 1a
- PV:
-
Parvalbumin
- RIPA:
-
Radioimmunoprecipitation assay buffer
- RT:
-
Room temperature
- STED:
-
Stimulated emission depletion
- TBS:
-
Tris buffered saline
- TDE:
-
2,2-Thiodiethanol
- VGAT:
-
Vesicular GABA transporter
- vGluT2:
-
Vesicular glutamate transporter 2
- WT:
-
Wild type
References
Tomchek SD, Dunn W. Sensory processing in children with and without autism: a comparative study using the short sensory profile. Am J Occup Ther. 2007;61(2):190–200.
Moore DJ. Acute pain experience in individuals with autism spectrum disorders: a review. Autism. 2015;19:387–99.
Duerden EG, Taylor MJ, Lee M, McGrath PA, Davis KD, Roberts SW. Decreased sensitivity to thermal stimuli in adolescents with autism spectrum disorder: relation to symptomatology and cognitive ability. J Pain. 2015;16(5):463–71.
Klintwall L, Holm A, Eriksson M, Carlsson LH, Olsson MB, Hedvall A, et al. Sensory abnormalities in autism. A brief report. Res Dev Disabil. 2011;32(2):795–800.
Tordjman S, Anderson GM, Botbol M, Brailly-Tabard S, Perez-Diaz F, Graignic R, et al. Pain reactivity and plasma β-endorphin in children and adolescents with autistic disorder. PLoS ONE. 2009;4(8):e5289.
Fründt O, Grashorn W, Schöttle D, Peiker I, David N, Engel AK, et al. Quantitative sensory testing in adults with autism spectrum disorders. J Autism Dev Disord. 2017;47(4):1183–92.
Riquelme I, Hatem SM, Montoya P. Abnormal pressure pain, touch sensitivity, proprioception, and manual dexterity in children with autism spectrum disorders. Neural Plast. 2016;2016:1723401.
Chen C, Hung AY, Fan YT, Tan S, Hong H, Cheng Y. Linkage between pain sensitivity and empathic response in adolescents with autism spectrum conditions and conduct disorder symptoms. Autism Res. 2017;10(2):267–75.
Cascio C, McGlone F, Folger S, Tannan V, Baranek G, Pelphrey KA, et al. Tactile perception in adults with autism: A multidimensional psychophysical study. J Autism Dev Disord. 2008;38(1):127–37.
Clarke C. Autism spectrum disorder and amplified pain. Case Rep Psychiatry. 2015;2015:1–4.
Loades ME. Evidence-based practice in the face of complexity and comorbidity: a case study of an adolescent with asperger’s syndrome, anxiety, depression, and chronic pain. J Child Adolesc Psychiatr Nurs. 2015;28(2):73–83.
Bursch B, Ingman K, Vitti L, Hyman P, Zeltzer LK. Chronic pain in individuals with previously undiagnosed autistic spectrum disorders. J Pain. 2004;5(5):290–5.
Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL, Nurnberger JI, Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18:362–76.
Han Q, Kim YH, Wang X, Liu D, Zhang Z-J, Bey AL, et al. SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron. 2016;92(6):1279–93.
Braz J, Solorzano C, Wang X, Basbaum A. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron. 2014;82(3):522–36.
Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. 2015;87(4):684–98.
Sheng M, Kim E. The Shank family of scaffold proteins. J Cell. 2000;113:1851–6.
Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–63.
Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486(7402):256–60.
Won H, Lee H-R, Gee HY, Mah W, Kim J-I, Lee J, et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486(7402):261–5.
Yoon S-Y, Kwon S-G, Kim YH, Yeo J-H, Ko H-G, Roh D-H, et al. A critical role of spinal Shank2 proteins in NMDA-induced pain hypersensitivity. Mol Pain. 2017;13:1744806916688902.
Pappas AL, Bey AL, Wang X, Rossi M, Kim YH, Yan H, et al. Deficiency of Shank2 causes mania-like behavior that responds to mood stabilizers. JCI insight. 2017;2(20):e92052.
Zeilhofer HU, Studler B, Arabadzisz D, Schweizer C, Ahmadi S, Layh B, et al. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J Comp Neurol. 2005;482(2):123–41.
Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, et al. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLOS Biol. 2005;3(5):0878–90.
Vong L, Ye C, Yang Z, Choi B, Chua S, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71(1):142–54.
Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CVE. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–34.
Bechara A, Laumonnerie C, Vilain N, Kratochwil CF, Cankovic V, Maiorano NA, et al. Hoxa2 selects barrelette neuron identity and connectivity in the mouse somatosensory brainstem. Cell Rep. 2015;13(4):783–97.
Hoshi M, Batourina E, Mendelsohn C, Jain S. Novel mechanisms of early upper and lower urinary tract patterning regulated by RetY1015 docking tyrosine in mice. Development. 2012;139(13):2405–15.
Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla KH, Sanmarti-Vila L, et al. Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J Neurosci. 1999;19(15):6506–18.
Boeckers TM, Liedtke T, Spilker C, Dresbach T, Bockmann J, Kreutz MR, et al. C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. J Neurochem. 2005;92(3):519–24.
Roselli F, Livrea P, Almeida OFX. CDK5 is essential for soluble amyloid β-induced degradation of GKAP and remodeling of the synaptic actin cytoskeleton. PLOS ONE. 2011;6(7):e23097.
Roselli F, Hutzler P, Wegerich Y, Livrea P, Almeida OFX. Disassembly of shank and homer synaptic clusters is driven by soluble b-amyloid 1–40 through divergent NMDAR-dependent signalling pathways. PLOS ONE. 2009;4(6):e6011.
Li S, Olde Heuvel F, Rehman R, Aousji O, Froehlich A, Li Z, et al. Interleukin-13 and its receptor are synaptic proteins involved in plasticity and neuroprotection. Nat Commun. 2023;14(1):200.
Saxena S, Roselli F, Singh K, Leptien K, Julien J-P, Gros-Louis F, et al. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80(1):80–96.
Li L, Rutlin M, Abraira VE, Cassidy C, Kus L, Gong S, et al. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell. 2011;147(7):1615–27.
Ouali Alami N, Schurr C, Olde Heuvel F, Tang L, Li Q, Tasdogan A, et al. NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018;37(16):e98697.
Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, et al. Structural and molecular interrogation of intact biological systems. Nature. 2013;497(7449):332–7.
Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14(1):22–9.
Olde Heuvel F, Holl S, Chandrasekar A, Li Z, Wang Y, Rehman R, et al. STAT6 mediates the effect of ethanol on neuroinflammatory response in TBI. Brain Behav Immun. 2019;81:228–46.
Osseforth C, Moffitt JR, Schermelleh L, Michaelis J. Simultaneous dual-color 3D STED microscopy. Opt Express. 2014;22(6):7028.
Wang X, Zhang J, Eberhart D, Urban R, Meda K, Solorzano C, et al. Excitatory superficial dorsal horn interneurons are functionally heterogeneous and required for the full behavioral expression of Pain and Itch. Neuron. 2013;78(2):312–24.
Lu J, Luo C, Bali KK, **e R-G, Mains RE, Eipper BA, et al. A role for Kalirin-7 in nociceptive sensitization via activity-dependent modulation of spinal synapses. Nat Commun. 2015;6:6820.
Roome RB, Bourojeni FB, Mona B, Rastegar-Pouyani S, Blain R, Dumouchel A, et al. Phox2a defines a developmental origin of the anterolateral system in mice and humans. Cell Rep. 2020;33(8):108425.
Maricich SM, Morrison KM, Mathes EL, Brewer BM. Rodents rely on Merkel cells for texture discrimination tasks. J Neurosci. 2012;32(10):3296–300.
Yalcin I, Charlet A, Freund-Mercier MJ, Barrot M, Poisbeau P. Differentiating thermal allodynia and hyperalgesia using dynamic hot and cold plate in rodents. J Pain. 2009;10(7):767–73.
Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KSR, et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science. 2005;307(5714):1468–72.
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63.
Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51(1):5–17.
Zylka MJ, Rice FL, Anderson DJ. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45(1):17–25.
Witschi R, Punnakkal P, Paul J, Walczak JS, Cervero F, Fritschy JM, et al. Presynaptic alpha2-GABAA receptors in primary afferent depolarization and spinal pain control. J Neurosci. 2011;31(22):8134–42.
Szabo NE, Da Silva RV, Sotocinal SG, Zeilhofer HU, Mogil JS, Kania A. Hoxb8 intersection defines a role for Lmx1b in excitatory dorsal horn neuron development, spinofugal connectivity, and nociception. J Neurosci. 2015;35(13):5233–46.
Eggeling C, Willig KI, Barrantes FJ. STED microscopy of living cells—new frontiers in membrane and neurobiology. J Neurochem. 2013;126:203–12.
Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, et al. Characterization of the Shank family of synaptic proteins Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem. 1999;274(41):29510–8.
Orefice LL, Mosko JR, Morency DT, Wells MF, Tasnim A, Mozeika SM, et al. Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cell. 2019;178(4):867–86.
Woelfle S, Deshpande D, Feldengut S, Roselli F, Deisseroth K, Michaelis J, et al. CLARITY increases sensitivity and specificity of fluorescence immunostaining in long-term archived human brain tissue. bioRxiv. 2022;2022.04.27.489700.
Rebelo S, Reguenga C, Lopes C, Lima D. Prrxl1 is required for the generation of a subset of nociceptive glutamatergic superficial spinal dorsal horn neurons. Dev Dyn. 2010;239(6):1684–94.
Hsu W, Mirando AJ, Yu HMI. Manipulating gene activity in Wnt1-expressing precursors of neural epithelial and neural crest cells. Dev Dyn. 2010;239(1):338–45.
Borromeo MD, Meredith DM, Castro DS, Chang JC, Tung KC, Guillemot F, et al. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development. 2014;141(14):2803–12.
Foster E, Wildner H, Tudeau L, Haueter S, Ralvenius WT, Jegen M, et al. Targeted ablation, silencing, and activation establish glycinergic dorsal horn neurons as key components of a spinal gate for pain and itch. Neuron. 2015;85(6):1289–304.
Alvarez FJ, Villalba RM, Zerda R, Schneider SP. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J Comp Neurol. 2004;472(3):257–80.
Bourane S, Duan B, Koch SC, Dalet A, Britz O, Garcia-Campmany L, et al. Gate control of mechanical itch by a subpopulation of spinal cord interneurons. Science. 2015;350(6260):550–4.
Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, et al. Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron. 2010;65(6):886–98.
Zeilhofer HU, Zeilhofer UB. Spinal dis-inhibition in inflammatory pain. Neurosci Lett. 2008;437(3):170–4.
Zeilhofer HU, Acuña MA, Gingras J, Yévenes GE. Glycine receptors and glycine transporters: Targets for novel analgesics? Cell Mol Life Sci. 2018;75(3):447–65.
Lu Y, Dong H, Gao Y, Gong Y, Ren Y, Gu N, et al. A feed-forward spinal cord glycinergic neural circuit gates mechanical allodynia. J Clin Invest. 2013;123(9):4050–62.
Vuilleumier PH, Fritsche R, Schliessbach J, Schmitt B, Arendt-Nielsen L, Zeilhofer HU, et al. Mutations affecting glycinergic neurotransmission in hyperekplexia increase pain sensitivity. Brain. 2018;141(1):63–71.
Zeilhofer HU, Neumann E, Munro G. Spinal GABAA receptors for pain control: Back to the future? Br J Anaesth. 2019;123(2):e176–9.
Boyle KA, Gradwell MA, Yasaka T, Dickie AC, Polgár E, Ganley RP, et al. Defining a spinal microcircuit that gates myelinated afferent input: implications for tactile allodynia. Cell Rep. 2019;28(2):526–40.
Petitjean H, Pawlowski SA, Fraine SL, Sharif B, Hamad D, Fatima T, et al. Dorsal horn parvalbumin neurons are gate-keepers of touch-evoked pain after nerve injury. Cell Rep. 2015;13(6):1246–57.
Ralvenius WT, Benke D, Acuña MA, Rudolph U, Zeilhofer HU. Analgesia and unwanted benzodiazepine effects in point-mutated mice expressing only one benzodiazepine-sensitive GABAA receptor subtype. Nat Commun. 2015;6:6803.
Ko HG, Oh SB, Zhuo M, Kaang BK. Reduced acute nociception and chronic pain in Shank2−/− mice. Mol Pain. 2016;12:1744806916647056.
Tsuda M, Koga K, Chen T, Zhuo M. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. J Neurochem. 2017;141(4):486–98.
Häring M, Zeisel A, Hochgerner H, Rinwa P, Jakobsson JET, Lönnerberg P, et al. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat Neurosci. 2018;21(6):869–80.
Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51(1):99–111.
Favaro PD, Huang X, Hosang L, Stodieck S, Cui L, Liu Y-Z, et al. An opposing function of paralogs in balancing developmental synapse maturation. PLOS Biol. 2018;16(12):e2006838.
Shi R, Redman P, Ghose D, Hwang H, Liu Y, Ren X, et al. Shank proteins differentially regulate synaptic transmission. Eneuro. 2017;4(6):ENEURO.0163-15.2017.
Monteiro P, Feng G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci. 2017;18(3):147–57.
Chung C, Ha S, Kang H, Lee J, Um SM, Yan H, et al. Early correction of N-Methyl-D-aspartate receptor function improves autistic-like social behaviors in adult Shank2−/− mice. Biol Psychiatry. 2019;85(7):534–43.
Coderre TJ, Vaccarino AL, Melzack R. Central nervous system plasticity in the tonic pain response to subcutaneous formalin injection. Brain Res. 1990;535(1):155–8.
Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2(12):1114–9.
Asante CO, Wallace VC, Dickenson AH. Formalin-induced behavioural hypersensitivity and neuronal hyperexcitability are mediated by rapid protein synthesis at the spinal level. Mol Pain. 2009;5:1744–8069.
Imlach WL, Bhola RF, Mohammadi SA, Christie MJ. Glycinergic dysfunction in a subpopulation of dorsal horn interneurons in a rat model of neuropathic pain. Sci Rep. 2016;6:1–14.
Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell. 2016;166(2):299–313.
Dawes JM, Weir GA, Middleton SJ, Patel R, Chisholm KI, Pettingill P, et al. Immune or genetic-mediated disruption of CASPR2 causes pain hypersensitivity due to enhanced primary afferent excitability. Neuron. 2018;97(4):806–22.
Loomes R, Hull L, Mandy WPL. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry. 2017;56(6):466–74.
Dedek A, Xu J, Lorenzo LÉ, Godin AG, Kandegedara CM, Glavina G, et al. Sexual dimorphism in a neuronal mechanism of spinal hyperexcitability across rodent and human models of pathological pain. Brain. 2022;145(3):1124–38.
Mogil JS. Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat Rev Neurosci. 2020;21(7):353–65.
Acknowledgements
The authors would like to thank prof. Frank Kirchoff for the use of the confocal facility and prof. Anita Ignatius for the access to the histology facility. The technical assistance provided by Thomas Lenk and Gizem Yartas is gratefully acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL. FR and TMB are supported by the DFG in the context of the CRC1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma” (DFG No. 251293561) and by the DFG with the Grant No. 431995586. FR and NOA is supported by the Baustein program of Ulm University. FR is also supported by the Synapsis Foundation, the Radala foundation and the Thierry Latran Foundation. EPZ is supported by the German Research Foundation (DFG, PO1319/3), the Federal Ministry of Education and Research (BMBF), the Federal Joint Committee (G-BA) and the Innovative Medicines Initiative 2 Joint Undertaking under grant Agreement No 777500. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA. EPZ also received financial support from Grunenthal for research activities and advisory and lecture fees from Grünenthal, Novartis and Medtronic. All money went to the institutions (WWU/UKM) EPZ is working for.
Author information
Authors and Affiliations
Contributions
FoH, NOA and OA, PZ and EPZ performed behaviour and histological analysis, conceptualization of the project and writing of manuscript. HW performed and analysed dorsal horn architecture. EK performed and analysed c-Fos in situ hybridization experiments. AC performed biochemistry on different brain regions and WT versus Shank2−/− samples. DD performed STED microscopy. SW, MS, SJ, SG provided various mouse lines or human material used in this study. ACL provided resources, reviewed, and edited the final draft. EPZ, PZ, CV and JM provided resources, assisted in conceptualization of the project, and reviewed the final draft. TMB and FR performed conceptualization of the project, resources, supervision, funding acquisition, project administration and writing and editing of the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experiments were performed in agreement with the local and national guidelines for animal experimentation. All experiments were approved by the Regierungspräsidium Tübingen under the licence n° o.103 and by the Italian Ministry of Health966/2016-PR.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
: Shank2 spinal cord localisation. Figure S2 Shank2 architecture and localisation. Figure S3 Loss of Shank2 immunoreactivity in cerebral cortex from Shank2−/− mice. Figure S4 CTB-488 injections reveal cutaneous afferents on Shank2high expressing neurons. Figure S5 No difference in neuronal architecture in the spinal cord of Shank2−/− mice. Figure S6 No difference in inhibitory synapses in the spinal cord of Shank2−/− mice. Figure S7 Increased number of cFos mRNA + interneurons in Laminae I upon Formalin injection. Table S1 Primary and secondary antibodies used for immunostainingFluorescent in Situ Hybridisationor for western blot. Table S2 Full details regarding human samples. Source data file 1 uncropped Western Blot of Shank2 in mouse tissue.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Olde Heuvel, F., Ouali Alami, N., Aousji, O. et al. Shank2 identifies a subset of glycinergic neurons involved in altered nociception in an autism model. Molecular Autism 14, 21 (2023). https://doi.org/10.1186/s13229-023-00552-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-023-00552-7