
Abstract
Primary liver cancer (PLC) is a common malignancy with high morbidity and mortality. Poor prognosis and easy recurrence on PLC patients calls for optimizations of the current conventional treatments and the exploration of novel therapeutic strategies. For most malignancies, including PLC, immune cells play crucial roles in regulating tumor microenvironments and specifically recognizing tumor cells. Therefore, cellular based immunotherapy has its instinctive advantages in PLC therapy as a novel therapeutic strategy. From the active and passive immune perspectives, we introduced the cellular based immunotherapies for PLC in this review, covering both the lymphoid and myeloid cells. Then we briefly review the combined cellular immunotherapeutic approaches and the existing obstacles for PLC treatment.
Similar content being viewed by others

Background
The twentieth century recorded increased cancer mortality rates, of which primary liver cancer (PLC), the fourth most lethal carcinoma, accounted for 8.2% of the total [1, 2]. The disease has the highest incidence rate in eastern Asia, and globally, every year, approximately 841,000 new cases and 782,000 deaths are recorded [1]. Therefore, PLC is a serious health and economic burden.
Histologically, PLC primarily comprises hepatocellular carcinoma (HCC) (75-85%), intrahepatic cholangiocarcinoma (iCCA) (10-15%) and other rare cases [2]. With complex etiological variation and occult clinical features, PLC is predominantly diagnosed at stages not early enough for simple surgical resection treatment, and patients experiencing high recurrence rates [3,4,5,6]. Targeted treatments based on natural or synthetic drugs revealed the positive antitumor effects against PLC [7,8,9,10]. Further researches have shown that combined therapeutic approaches, including interventional therapy, radiotherapy, chemotherapy and biotherapy, improve the curative effects and the possibility for individual treatment in PLC [11]. HCCs and iCCAs potentially share the common hepatocyte origins despite their histologically distinctive clinical features, and a final tumor phenotype could be affected by interactions between the immune microenvironment and oncogenes [12]. Immune surveillance, whereby tumor cells are eliminated at nascent stages, protects the body from tumors. Using antigenic modulation, tumor-derived soluble factors, and immunological ignorance and tolerance strategies, tumor cells become capable to survive from the host’s immune attack, and homeostasis gradually proceeds from immune surveillance to equilibrium and further immune escape during tumor progression [13]. These interactive procedures suggest immunological intervention may have potential to limit or even reverse this phenotypic transformation, under certain conditions.
By modifying the immune system to elicit antitumor capabilities, immunotherapies are viable alternatives for PLC therapy. These aforementioned immunotherapeutic strategies can be categorized into active and passive immunotherapy according to the immune response mechanisms. Immune cells play crucial roles in the antitumor procedure constructed by immune system both in innate and adaptive immunity, thus cellular based immunotherapy has underpinned numerous immunotherapeutic approaches for PLC currently. Based on the means of interventions, the cellular immunotherapies are typically composed of active and passive immune therapeutic strategies. In this review, we introduce the cellular based immunotherapeutic approaches for PLC (Fig. 1), with a brief overview of combined cellular treatments and current therapeutic issues.

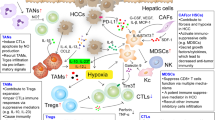
Cellular based immunotherapy in liver cancer. Based on myeloid or lymphoid immune cells, strategies are attempted for liver cancer therapy. In myeloid cell group, DC vaccine, engineered Mφ and depletion of immune suppressors are undergoing research for hepatoma treatment. For lymphoid cells, strategies such as T/NK cell engineering, Tregs/Bregs depletion and molecular regulatory intervenes are also under study. DC, dendritic cell; Mφ, macrophage; Tregs, regulatory T cells; Bregs, regulatory B cells; FOXP3, forkhead box protein P3; GITRL, ligand to Tregs evoked glucocorticoid induced tumor necrosis factor receptor; TCR-T, T cell receptor engineered T cells; CAR-T, chimeric antibody receptor engineered T Cells; HBV, hepatitis B virus; HCV, hepatitis C virus; HLA-A2, human leukocyte antigen-A2; AFP, A-fetoprotein; GPC3, Glypican-3; NKG2D, NK group 2 member D; VEGF, vascular endothelial growth factor; EGFRvIII, epidermal growth factor receptor variant III; TIM-1+, T cell immunoglobulin mucin domain-1 positive; PD-1, programmed cell death-1; CXCL9, chemokine C-X-C motif chemokine ligand-9; IL, interleukin; CD169, cluster of differentiation 169; CD44, cluster of differentiation 44; CD133, cluster of differentiation 133; CD40, cluster of differentiation 40; CD160, cluster of differentiation 160; CD96, cluster of differentiation 96; CD11b, cluster of differentiation 11b; CD27, cluster of differentiation 27; CD3, cluster of differentiation 3; DAP10, DNAX-activating protein 10
Active cellular immunotherapy in primary liver cancer
Cell vaccines
Vaccine treatments are based on tumor antigens which activate the host’s immune system to eliminate tumor cells and memorize abnormal antigens for tumor recurrence. Aimed at evoking immune response to tumor-specific/associated antigens (TSAs/TAAs), exogenous vectors, intracellular elements (peptides, proteins or nucleic acids), and correlated cells have been proposed as antitumor vaccines [14]. Cell vaccines, including allogeneic and autologous groups [15], arise several approaches for PLC therapy. When compared with viral, bacterial or yeast vectors, cell vaccines are advantageous of avoiding the immune responses triggered by exogenous vector carriers.
Allogeneic cell vaccines are usually prepared from tumor cells or lysate collections, with or without gene modification, before final TSA/TAA delivery to the immune system. The approach is advantageous as reagents can be mass produced but is flawed in terms of maturation for antigen presenting cells (APCs) [15, 16]. In vitro evidence suggested that the allogeneic cell lines, HepG2 and BEL7402, when co-cultured with autologous dendritic cells (DCs), the functional APCs in the body, emerged a positive activation of both CD4+ and CD8+ T cells against autologous hepatoma cells [17, 18]. An in vitro iCCA study revealed similar results to HCC: RNA and protein lysates extracted from iCCA cell lines have talents to pulse DCs and enhance T cell cytotoxicity against cholangiocarcinoma [19]. Clinical trials on allogeneic cell vaccines against PLC are also underway (Table 1) (clinicaltrials.gov). Based on antitumor immunity in animal models induced by allogeneic cancer stem cell vaccination [28], further clinical trials against HCC have been completed and awaiting results (NCT02089919). The phase I clinical trial of ilixadencel [20], an allogeneic DC vaccine, confirmed its safety and effectiveness in activating tumor specific immune responses in advanced HCC (NCT01974661). However, more in-depth investigations are required to apply these therapies to PLC in clinical settings.
Autologous cell vaccines, which present effective TSAs/TAAs, are derived from and returned to patients after in vitro manipulation [15]. Both tumor cell based and APC based autologous vaccines have displayed anticancer potential towards PLC in recent studies (Table 1). The safety of hybrid cell vaccination was certified in liver involved metastatic melanoma [29]. The Hepa 1-6 cell vaccine, equipped with granulocyte macrophage colony stimulating factor (GM-CSF) and interleukin-2 (IL-2) as adjuvants, was protective against HCC in a syngeneic C67L/J mouse model, and the autologous fixed tumor formulation vaccine was validated as preventing HCC recurrence in phase I/II clinical trials [21, 22]. In other research, a bi-shRNAfurin/GM-CSF incorporated autologous HCC cell vaccine, FANG™, stabilized PLC for over 4 months in five patients during a phase I trial, with four patients experiencing more than 2 years’ survival, which surpassed the 7.9 month median survival rate of sorafenib in a phase III trial [23, 30]. Autologous DC-tumor vaccines have also shown safety and protective effects from recurrence and metastasis for postoperative HCC [24]. The safety of tumor lysate pulsed DC vaccines for PLC was tentatively confirmed in other clinical trials, while boosters following DC therapy showed increased efficacy in prolonging survival for HCC patients when compared with single pulsed DC vaccines [25, 31]. A previous study showed that iCCA cell lysate pulsed autologous DCs, especially gene modified self-DCs, enhanced effector T cell cytotoxicity against iCCA [26]. A 6-year follow up based clinical trial on appraising the positive effects of tumor lysates pulsed DC vaccine for iCCA also demonstrated the feasibility and effectiveness (UMIN000005820) [27]. Preliminary findings on autologous cell vaccine for PLC therapy showed positive achieves, however, further investigations are still needed to better understanding the underlying mechanisms for clinical applications.
Negative lymphoid regulatory cell blockage
Lymphoid regulatory cells participate in monitoring internal immune homeostasis, the negative functional ones including FOXP3+CD25+CD4+ regulatory T cells (Treg) and regulatory B cells (Breg) possess inhibitory roles on antitumor immunity in liver cancer [32,33,34]. Strategies to block regulatory cell mediated immunosuppression, either by depleting effector regulatory cells or modulating correlated activating pathways, may play crucial roles in achieving immunotherapy against liver cancer.
Tregs are broadly classified into thymus-derived naturally occurring Tregs (tTreg cells) and peripherally derived induced Tregs (pTreg cells), cohesively regulating internal immune homeostasis [35, 36]. Tregs suppress APC function via down-regulating CD80/CD86 with cytotoxic T lymphocyte associated antigen-4 (CTLA-4) expression, and reduce responder T cells by competitively consuming surrounding interleukin-2 (IL-2), as key mechanisms in cancer immune suppression, proceed effector T cell anergy in antitumor response [36]. The immune suppressive modulation of singularly recruited Tregs in PLC has been validated both in vitro and vivo studies [32, 37,38,39,40]. Currently, Tregs blunt antitumor immunity via immune cell correlated intervening approaches in PLC (Fig. 2): 1) APC suppression: Tregs harvested from HCC mice inhibit DC function by down-regulating the co-stimulator CD80/86 via CTLA-4 expression, secreting inhibitory cytokines such as IL-10 to weaken DC maturation and the tumor necrosis factor-α (TNF-α)/IL-12 production, and inhibiting via cell to cell contacts [41]. The toll like receptor-4 (TLR-4) triggers interactions between Tregs and macrophages, leading to immune suppression in HCC [42]. 2) FOXP3− T cell suppression: Tregs attributed to the programmed cell death-1 (PD-1) correlated dysregulation of T cell population frequencies, with the exhaustion of functional T cells [40, 43]. Cytokines like IL-2 with its highly expressed CD25 receptor on Tregs may also play critical role in functional T cell toxicity [44, 45]. Tregs impair γδ T cells and down-regulate the interferon-γ (IFN-γ) secretion of γδ T cells in a transforming growth factor β (TGF-β) and IL-10 dependent manner [37]. Previously, the deletion of tumor infiltrating Tregs was demonstrated to enhance HCC specific immunotherapy [46]. Drugs such as solanine (CD4+CD25+ Tregs’ proportion decreaser) [47] and astragalus polysaccharides (FOXP3 expression inhibitor) [48] also revealed antitumor enhancement in HCC via Treg suppression. Based on predecessor’s work, attempts on rectifying Tregs mediated immune dysregulation in PLC therapy are never stagnation. Tregs evoked glucocorticoid ligands induced tumor necrosis factor receptor expression in PLC, and are proposed as potential treatments for PLC by decreasing Treg immunosuppression and reactivating CD4+CD25− T cells [32]. Furthermore, when combined with a CTLA-4 blockade, this was shown to improve antitumor efficacy during treatment [49]. For patients resistant to immune checkpoint inhibitor treatment, cabozantinib, exerted its immune regulation effects via releasing HGF (hepatocyte growth factor) correlated DCs suppression and Tregs promotion, is currently being explored in a phase III clinical trial to verify its therapeutic capacity for HCC (NCT04588051). Further exploration for the Treg based effector mechanisms and therapeutic methods are required in PLC.

Tregs interact with immune cells and the therapies in liver cancer. Tregs suppress anti-liver cancer immunity via interacting with several immune cells. Firstly, Tregs inhibit APCs’ function in liver cancer, currently known mechanisms like CTLA-4 ligand expression to down-regulate DCs’ CD80/86 and IL-10 secretion to inhibit DCs maturation, TLR-4 signal mediated immune suppression with macrophage participant. And the APCs suppression may be rescued by drugs like cabozantinib and CTLA4 blockade. Secondly, Tregs suppress FOXP3− T cells in liver cancer such as effector T cell (consuming IL-2 with highly expressed CD25; PD-1 correlated dysregulation) and γδ T cell (depending on TGF β and IL-10), which can be partially blocked by GITRL therapy. And regulatory T cell itself can be depressed by CD4+CD25+ Tregs’ proportion decreaser solanine and FOXP3 expression inhibitor astragalus polysaccharides for liver cancer therapy. CTLA-4, cytotoxic T lymphocyte associated antigen-4; IL-2, interleukin-2; IL-10, interleukin-10; TLR-4, Toll like receptor-4; PD-1, programmed cell death-1; PD-L1, programmed cell death-ligand 1; TGF β, transforming growth factor β; GITRL, glucocorticoid induced tumor necrosis factor receptor ligand; CD25, cluster of differentiation 25
Bregs, differentiated from B lymphoid cells, were demonstrated to play pivotal roles in anti-immune response activity against tumor, while their surface tags for phenotype classification have not yet reach consensus so far [50, 51]. To elicit immune suppressive efficacy during tumorigenesis, Bregs function in diverse differentiation and functional mechanism in immune cells, such as IL-10 dependent inhibition on APCs and cytotoxic T cells, and the cytokine (IL-10, TGF-β) relied promotion of FOXP3+ T cell differentiation in the immune system [50].
A high Breg frequency was correlated with HCC in rats [52], the correlation was also supported by the raising frequency for Bregs in postoperative HCC patients [34]. To our knowledge, Bregs interact with PD-L1 and lead to T cell dysfunction in an IL-10 dependent manner in hepatoma [53], They also accelerate the proliferation and invasion of HCC cells via the CD40/CD154 pathway, and the Breg frequency was positively relating to advanced HCC stages [33].
Increased TIM − 1+ Breg cell frequency was closely associated with HCC malignant progression and poor prognosis, which evidently proved to be mediated by tumor sourced high mobility group box 1 (HMGB1) via toll like receptor 2/4 (TLR2/4)-mitogen activated protein kinase (MAPK) pathway. Together with anti-PD-1/PD-L1 therapy against PD-1hiBregs, therapies targeted at Breg specific tags enlighten potential blockage paths against immune escape in HCC [53,54,55]. Trials on Bregs targeted therapy, like Total glucoside of paeony, was also confirmed with efficiency in HCC rats [56]. However, to fill the gaps between theoretical researches and clinical practices of Bregs targeted treatment against PLC, further explorations on the correlation between Bregs and liver cancer are still needed.
Passive cellular immunotherapy in primary liver cancer
Lymphoid cell based immunotherapy for primary liver cancer
Engineering T lymphocytes for primary liver cancer therapy
Originating from myeloid lymphatic stem cells and matured in thymus, T cells participate in many aspects of acquired immunity, both cellular immunity and a lesser extent to humoral immunity, to maintain homoeostasis. CD3−CD4−CD8− triple negative T cells, namely bone marrow T progenitors, are selected and rearranged for specific T cell receptor (TCR), then differentiate into CD3+CD4+CD8− αβTCR helper T cells (CD4+ T), CD3+CD4−CD8+ αβTCR cytotoxic T cells (CD8+ T) and CD3+CD4−CD8− γδTCR T cells (γδ T) for immune function by thymopoiesis [57]. Dysfunctions of T cells including cell repertoire distributional aberrance, transcriptional regulation and pathway regulative changes, were found to be liable for various kinds of tumorigenesis, and higher proportion of CD3+ or CD8+ T cells were validated to have correlation with better clinical outcomes in both HCC and iCCA [58,59,60], therefore T cell targeted intervenes are concerned as potential immune therapeutic strategies for PLC. Gene modified therapy, such as retrovirus transduced epidermal growth factor receptor (EGFR) expressing CD8+ T cell, was validated of having tumor growth suppression efficiency in mice [61]. Targeted at blocking up CD8+ T cell exhaustion correlated negative costimulatory molecules, immune checkpoint blockages such as tremelimumab, pembrolizumab, nivolumab and ipilimumab are undergoing clinical trials for HCC therapy [62], and etiology specific immunotherapies potentially elicit better outcomes [63]. Clinical case reported that allogenic γδ T therapy enhanced the peripheral immune response against iCCA and improved the patient’s prognosis (NCT02425735) [64].
Among multifarious immunotherapies, genetically TCR engineered T cells (TCR-T) and chimeric antibody receptor engineered T Cells (CAR-T) are pioneer and efficient attempts for the application of engineered T cells in adoptive cellular therapy for PLC. To evaluate the safety and efficiency of TCR-T/CAR-T for solid malignancies, including hepatoma, phase I/II clinical trials are projected and under recruiting (NCT03941626, NCT03638206). Compared with CAR-T, TCR-T is deficient in major histocompatibility complex (MHC) restriction on recognizing TSAs/TAAs but have broader scope on recognizing tumor intracellular proteins, which makes it much advantageous on solid tumor therapy [65]. A-fetoprotein (AFP) specific CD8+ T cell clusters, deprived from human leukocyte antigen (HLA)-A2 transgenic AAD mice, were hybridized to generate CD8+ T cell with HLA-A2/AFP identifiable TCR, and the hybridoma T cell clones were detected to have effective toxicity on HCC tumor cells [66]. The immune therapeutic potency of HLA-2/AFP specific TCR against HCC was also confirmed with human peripheral blood mononuclear cell (PBMC) derived CD8+ T cells [67]. Further trials for AFP specific TCR T cells used on clinical therapy are underway (Table. 2). C-TCR055, AFP specific TCR T cell injection, was selected for function and safety from TCR profiles, and has been used to initiate phase I clinical trials for unresectable HCC therapy (NCT03971747, NCT04368182) [73]. Autologous genetically modified AFPc332T cells used for therapeutic trial on advanced HCC are under recruiting (NCT03132792). High affinity purposed TCR engineering, targeted at hepatitis B virus (HBV) [74] and hepatitis C virus (HCV) [75] improved the sensitivity and cytotoxicity of T cell therapy for virus related HCC. HBV specific TCR engineered T cells exhibited cytotoxicity against HBV DNA naturally integrated HCC cells in vitro [76]. Phase I clinical trials tentatively verified that autologous HBV-TCR T cell therapy decreased the pulmonary metastases of HCC free for affecting liver function [68], and provide valuable prevention against HCC relapse with at least 4 weeks’ post-transfer exhibition in patient [69]. New approaches for TSAs/TAAs targeted CAR-T are also springing up in PLC immunotherapy. Glypican-3 (GPC3) targeted CAR-T therapy, such as G3-28Z-41BBL CAR-T [77] and 32A9 CAR-T [78], displayed the cytolytic activities against GPC3+ HCC cells. Further optimizations on GPC3-CAR-T positively support its application in HCC treatment. For example, co-expression with IL15/IL21 expanded the antitumor activity of GPC3-CAR-T against HCC at laboratory level [79]. The modification of C-X-C motif chemokine receptor 2 (CXCR2) expression to GPC3-CAR-T promoted its migration and cytotoxicity against HCC cells in mice [80]. The combination with subpharmacologic dose of sorafenib enhanced the antitumor efficiency of GPC3-CAR-T in HCC mouse model [81]. The split GPC3-CAR-T suppressed tumor growth also reduced the risk of severe cytokine release syndrome in vitro and xenograft mice model [82]. GPC3-CAR-T also showed tumor eliminating capabilities in HCC patient derived xenograft (PDX) models (NCT03198546) as a potential CAR-T candidate for PLC therapy [70]. The efficacy, safety and pharmacological properties of GPC3-CAR-T are undergoing clinical trials for further verification (NCT04121273, NCT03884751, NCT03302403, NCT03980288, NCT02905188). Gene modifications for CARs have been constructed to recognize abnormally expressed antigens in malignant cells. The NK group 2 member D (NKG2D) ligands (NKG2DL), highly expressed in tumor cells, were tested as CAR-T targets, and showed that NKG2D-based CAR-T effectively kill NKG2Dhigh HCC cells [83]. Intratumoral medication of AFP-CAR-T was reported to lyse HCC cells via cytokine dependent manner and suppress tumor growth in mouse model [84]. Analogously, CD44-CAR-T [85] and EGFRvIII-CAR-T [86] both released higher levels of cytokines such as INF-γ, TNF-α and better suppressed HCC growth compared with normal/mock T group in vitro and vivo. In a phase II clinical trial, patients with advanced HCC received CD133-CAR-T cell infusion after prior systemic therapy, and emerged with 12 months median overall survival (OS) and 6.8 months progression free survival (PFS) [71]. These observations were correlated with significantly increased vascular endothelial growth factor (VEGF) and stromal cell derived factor 1 (SDF-1) levels (both positive for longer OS and PFS), and decreased endothelial progenitor cell (EPC) levels (positive to shorter OS) (NCT02541370). Cocktail treatment, comprised with VEGF-CAR-T, PD-1 monoclonal antibody and CD133-CAR-T, was reported to be effective for iCCA, with a clinical case showed the patient acquired a total of 13-month partial response (PR) from CAR-T therapy, while the toxicities need further exploration [72]. More comprehensive engineered T cell therapy studies are required for further clinical applications.
B lymphocytes targeted strategies for liver cancer therapy
B cells originate from lymphoid stem cells and develop into functional subgroups, such as CD5+B-1 for inherent immunity, CD5−B-2 for adaptive humoral immunity, and Bregs for immune suppression [87]. B cell dysregulation, such as metabolic dysfunction and subset distribution derangement, may contribute to oncogenesis, therefore therapeutic strategies targeted at correcting dysregulations in B cells are likely to generate beneficial antitumor immunity [88, 89]. Patients with type II diabetes were sighted of high immature/transitional B cell frequencies, which might be liable for the procession of chronic hepatitis C (CHC) to HCC and considered as potential disease predictors for CHC [89]. The correlation between B cell dysregulations, either the metabolic changes or subsets redistributions, and the tumorigenesis of PLC are less clear so far. Elimination of CD20+B cells with CD4+/CD8+ T reserved showed inhibition effects on liver cancer progression in Mdr2−/− mice under liver fibrosis condition [90], while clinical studies revealed that B cells were notably decreased in HCC, and the density of tumor infiltrating CD20+B cells was positively correlated with superior survival as well as CD3+T cells [91, 92]. Further investigations on interactions between tumor infiltrating B cells and T cells, and to verify whether a compensatory mechanism or species variation exists are needed. CD40, a member of TNF receptors, is broadly expressed on immune cells like DCs, B cells as well as some tumor cells. The agonistic reagents to CD40 showed the activation impacts on antitumor immunity as immunotherapeutic candidates [93]. Compared to single monoclonal antibody (mAb) or chemotherapy groups, the combination of anti-CD40/PD-1 with chemotherapy significantly impaired tumor growth and prolonged survival in advanced iCCA murine model [94]. While study also suggested that agonistic anti-CD40 may impel the maturity of myeloid suppressive cells and result in liver damage in mice [95]. Clinical trial to evaluate the efficiency and tolerability of CD40 antibody CDX-1140 in advanced malignancies including PLC is under recruiting for next step estimation on CD40 antagonists (NCT03329950). In depth studies on B cell dependent therapies for PLC and associated mechanisms are warranted in next stage.
Natural killer cell based intervetions for liver cancer therapy
Hematopoietic stem cells derived Natural killer (NK) cells are CD3− lymphocytes which classified as minor excretive CD56bright or major cytocidal CD56dim subsets. NK cells play important parts in innate immunity, regulatory immunity, also protect the body from tumor, virus and parasitic bacterium with no prior antigen sensitization requirement [96]. NK cell abnormality is correlated with immunologic defect, as possible causative to liver disease including viral hepatitis, autoimmune disease and liver cancer [96, 97]. For feedback, tumor microenvironments also have impacts on regulating the function and collaboration of NK cells with other immune cells in PLC [98]. NK phenotype has positive or negative effects on HCC in clinical observations, differently. Study showed that the decreasing of CD160+NK cells in intra-HCC tissue lead to worsened disease progression with higher recurrence rates, whereas TGF-β1 blocking intervene can restore the CD160+NK cell proportion [99]. CD96+ NK cells were notably increased in HCC tumor tissue and linked to poor clinical outcomes, while the blockage of TGF-β1 or CD96-CD155 interaction can rescue the NK cell dysfunction and proposed possible routes for PLC therapy [100]. Similarly, CD11b−CD27−NK cells were highly infiltrated in tumor tissue of HCC patients, positively correlated with tumor progression and poor prognosis [101].
Strategies targeted at NK cell modulation, such as cytokine intervened and gene modified adoptive NK cell transfer, are forefront attempts for liver cancer [102,103,104,105,106]. IL1α was detected to have promotional effects on the cytotoxicity of NK cells against HCC [107]. IL-12/15/18 trafficked to spontaneous HCC mice model were also found to activate NK cells and lower tumor formation [105]. In iCCA, higher expression level of IFN-γ inducible chemokine C-X-C motif chemokine ligand-9 (CXCL9) was correlated with larger tumor infiltrating NK cells and longer postoperative survival [108]. Gene modifications on NK cells, like CAR-NK proposed their therapeutic potency against liver cancer in laboratory level. GPC3-CAR significantly enhanced the cytotoxicity and cytokine production of NK cells when co-cultured with GPC3+ HCC cells [104]. Similarly, cytotoxicity against liver cancer presented expanded effects with NKG2D-CD3ζ-DAP10-CAR activated NK cells [103] and hIFN-α transferred NK cell lines [106]. Strategies such as iconographic guidance [109] and carrier optimization [110] for NK cell used in liver cancer therapy also showed preferable antitumor efficiency, but still warrant clinical verifications. Clinical trials on allogeneic NK (NCT03937895, NCT03358849, NCT04162158, NCT02562963), CAR-NK adoptive immunotherapy (NCT02839954) to confirm the efficiency, safety and recurrence prevention role against PLC are awaiting for results.
Myeloid cell based immunotherapy for primary liver cancer
Monocyte/macrophage and liver cancer immunotherapy
Monocytes are generated from myeloid progenitors in bone marrow and released into circulation, then shaped into different phenotypes at specific tissue microenvironment, and can also polarize to replenish the innate macrophages [111,112,113]. Monocytes and macrophages were reported to have correlation with the progression of PLC [114,115,116], from which, the idiographic functional pathways may provide optional target molecules for liver cancer immunotherapy.
High monocyte counts predict worse prognosis for postoperative HCC patients, especially the hepatitis virus B infected group [117]. Low lymphocyte to monocyte ratio is companied with inferior HCC outcomes, with cirrhosis arising, total bilirubin elevation, tumor size enlargement and overall survival reduction [118]. Cohort observation on the linkage between lymphocyte monocyte ratio and HCC outcome has also been designed for clinical trial (NCT03869151). Monocyte subgroups were found to promote liver carcinogenesis by complex interactions with immune cells and particular molecules. Studies showed that hepatic stellate cells shift the monocyte into immunosuppressive phenotypes, namely kinds of myeloid derived suppressor cells (MDSCs), may promote liver injury as well as HCC recurrence and progression [119, 120]. Monocytes suppressed the cytotoxicity of retroviral transduced TCR T cells against hepatitis B virus related HCC via PD-1/PDL-1 signaling in a 3D model [121]. Peritumoral monocytes/macrophages were found to have correlation with intratumoral NK dysfunction via blocking CD48 protein 2B4 receptor on NK cells in advanced HCC [98]. Peritumoral monocytes also induced tumor cell autophagy to invade its edge and allow tumor metastasis in HCC [122]. TIE-2 expressing monocytes (TEMs) were positively correlated with HCC angiogenesis [123] and advanced disease stages with sorafenib therapy, emerging its potency as a novel marker in HCC [124, 125]. Ly6C+monocytes augment the myc triggered carcinogenesis and liver injury, while the tumor burden and survival of mice model can be rescued after monocytes depletion [114]. In spontaneous HCC mouse model, the deletion of IL-6 in monocytes/kupffer cells resulted in tumor suppression for optional therapeutic object [126]. These cell interaction networks and specific molecules suggest optimizational targets for PLC immunotherapy. Hepatic macrophages play crucial roles in hepatocarcinogenesis, both the positive and negative side with signal induced differential phenotypes to our knowledge, such as negatively releasing tumor promoting cytokines in pre-metastatic niche formation and extravasation, positively eliminating hepatoma cells by phagocytosis in cancer cell arrival stage [127,128,129,130]. CD169+macrophage subpopulations were found to enhance the cytotoxicity and amplification capability of CD8+T cells against HCC under anti-CD3 irritation, and exerted suppressive effects on tumor progression [131]. M2 macrophages were stimulated by hepatoma cells and result in tumor growth and metastases for both HCC and iCCA, therefore the blockage at correlated key molecules could serve as beneficial immunotherapeutic strategies in PLC [115, 132, 133]. M1 macrophage loaded hydrogel treatment significantly accelerated HCC tumor necrosis and decreased the tumor size in mice model [134]. Clinical trial on CAR-macrophages for HER2 overexpressing solid tumors, including HCC, is under recruiting (NCT04660929). For the conversion from laboratory research to patient therapy in next stage need more dependable evidence.
Monocytic MDSCs (M-MDSCs), similar to monocytes in morphology and phenotype, are more immunologic suppressive than the other polymorphonuclear MDSCs (PMN-MDSCs) branch in tumor tissue, with an alternative differentiation to tumor associated macrophages (TAM) other than mature macrophages and DCs [135]. Clinical lines showed that the frequency of M-MDSCs and total MDSCs was positively related with alanine transaminase (ALT), AFP, and HCV viral load, while presented negative correlation with CD8+ T cell frequency in HCV-HCC patients [136]. Indeed, study also reported that tumor associated fibroblasts (TAFs) treated monocytes, resembling to CD11b+ myeloid cells, possess impairments on T cells which negatively correlated with HCC progression [137]. Thus, M-MDSC targeted therapies have caught the attention of researchers and achieved several progresses in HCC treatment. In fibrotic livers, hepatic stellate cell induced increasing of M-MDSCs was found to promote HCC growth in both patients and mice model via p38 MAPK signalling, while status can be rescued by molecular targeting blockage on this pathway [138]. A traditional chinese decoction therapy of jianpi huayu showed its regulatory potency on facilitating the differentiation of MDSCs into macrophages and DCs in HCC mice model, and alleviated the immunosuppression on CD4+ T cells, which unfolded new perspectives on DCs/MDSCs targeted treatment against PLC [139].
Granulocytes and liver cancer immunotherapy
Granulocytes, mainly composed of neutrophils, eosinophils and basophils, are important components of myeloid cells, which exhibit characterized heterogeneity in inflammation and tumorigenesis [140, 141]. Granulocytic myeloid derived suppressor cells were demonstrated to rescue the blockage of tumor associated macrophages and promote iCCA progression [142]. Neutrophils play crucial roles in tumor mocroenvironment, and were reported to have close correlation with PLC progression [143,144,145]. High neutrophil counts revealed the predictive capacity to inferior clinical outcomes [145]. The neutrophil to lymphocyte ratio were also positively associated with HCC malignancy, like tumor aggression, extrahepatic recurrence and shrunken overall survival [146, 147]. The neutrophil extracellular traps were found to have tumor promoting effects on nonalcoholic steatohepatitis [144], suggesting that rational elimination or blockage on neutrophils could generate pleasant antitumor effects. Tumor associated neutrophils (TANs) triggered HCC cells and the initiated positive feedback loop for more TANs recruitment result in tumor progression [148]. TANs also recruited both macrophages and Tregs, leading to promote tumor growth and resistance to sorafenib [149]. Granulocytes targeted therapeutic strategies may be effective against PLC, however, more supportive evidence is required.
Morphologically and phenotypically more like neutrophils, PMN-MDSCs present relatively mild immunosuppressive effects but mainly work on regulating tumor specific immune responses, take the dominant place over M-MDSCs in peripheral lymphoid organs [135]. High level of LOX-1+ CD15+ PMN-MDSCs was proved to have correlation with poor prognosis in HCC patients via T cell suppression, which provided possibilities for PMN-MDSCs targeted therapy in liver cancer [150]. Cell cycle-related kinase (CCRK) depletion leaded suppression of PMN-MDSCs also displayed enhancement on the intratumorous CD8+T cells and PD-L1 blockade efficiency against HCC at laboratory level [151]. Further researches are needed to mature the theoretical and practical guideline of PMN-MDSCs targeted treatment against PLC.
Cellular immunotherapy combination in primary liver cancer
Treatment options for PLC are typically tailored to disease stages. At early stages of PLC, patients would be appropriate for surgical resection or liver transplantation under certain indications, combined with proper adjuvant therapies, such as Transarterial embolization/TAE, transcatheter arterial chemoembolization/TACE and radiofrequency ablation, to reduce recurrence. For advanced PLC, expectant systemic treatments like cytotoxic chemotherapy, oncolytic virus therapy and immunotherapy are better recommended for patients [11]. Comprehensive therapeutic projects, whether combined with conventional or novel strategies, revealed their superior curative effects against PLC.
Cellular immunotherapy combined with interventional treatment, targeted treatment and radiotherapy were found to have optimized curative effects against PLC. An open label clinical trial enrolled 52 participants on accessing the efficiency and safety of combined treatment against HCC, which composed of TACE and central memory T cells, is completed and awaiting results (NCT03575806). A combination of allogenic NK cell therapy notably increased the median overall survival for patients to 10.1 months, presented synergistic efficiency with irreversible electroporation (IRE) when performed for stage IV HCC [152]. DC-cytokine induced killer (CIK) treatment was detected to improve the antitumor efficiency against liver cancer in rats [153]. Further clinical research showed that a combination of DC-CIK with cryoablation treatment prolong the median overall survival of patients with metastatic HCC compared to single treated groups [154]. In phase I clinical trial, percutaneous microwave ablation prescribed with tumor lysate pulsed DCs, DC-CIK and cytotoxic T lymphocytes showed no adverse effects in HCC patients, and presented effector T cells increasing with Tregs decreasing 1 month after treatment [155]. A combination of toll like receptor-9 agonist and radiofrequency ablation better activated the peripheral blood mononuclear cells against VX2 hepatoma compared with single radiofrequency ablation, increased the antitumor effects and prolonged the survival in VX2 rabbit model [156]. Targeted treatment for blocking tumor progression, either with or without combination to cellular immunotherapy, showed remarkable therapeutic efficiency against PLC. Sorafenib, a multikinase inhibitor, was found to improve the antitumor efficiency in HCC mice model when combined with GPC3-CAR-T [81], and it also ameliorated the outcome of HCC patients when combined with NK cells [157]. AFP specific ET140202-T cells combined with sorafenib or TAE therapy against PLC are under recruiting for a phase I clinical trial (NCT03965546). As fibroblastic growth factor (FGF) signaling was detected to take on crucial parts of cellular characteristics in tumorigenesis, the blocking-up at fibroblastic growth factor receptor (FGFR) has also been focused, such as pan-FGFR inhibitors, and indeed showed its antitumor efficiency in PLC [158,159,160,161,162]. Infigratinib, a pan-FGFR inhibitor, was found to suppress the tumor growth of FGFRhi HCCs possessing sorafenib resistant, and improve the antineoplastic efficiency against HCC either combined with vinorelbine or bevacizumab [163, 164]. Supportively, FGFR mutations are reported to be correlated with indolent iCCA progression [165]. The abundant correlations between FGF signaling and immune cells, known like cell polarization [166] and metabolic regulation [167,168,169] to macrophage/monocyte, chemotaxis promotion for neutrophil [170], functional cytokine secretory regulation of B cells [171], also provide expectable possibilities for therapeutic combinations of FGFR inhibitors and immune cells against PLC. Lenvatinib, a multi targets inhibitor including FGFR, was proved to prolong the post progression survival of patients with unresectable HCC, and slightly increase the patients’ overall survival compared to first-line sorafenib [172, 173]. Case of iCCA also reported that lenvatinib suppress the metastasis progression when combined with nivolumab, an anti-PD-1 agent, which prompt novel perspectives on FGFR combined immunotherapy by clinic [174]. At laboratory level, Lenvatinib plus PD-1 blockade therapy was found to enhance the potency of effector T cells adjoint with decrease of monocytes and tumor associated macrophages, thus reach a preponderant antitumor effectiveness [175,176,177]. Further studies are required to insight the combination of immmue cells with multi kinase inhibitors used in PLC therapy. Radiotherapies, such as 125I joined with CIK, played inhibitory role on tumor growth in HCC mouse model, and showed improved outcomes [178]. 125I combined with NK cell therapy reportedly enhanced immune responses and reduced tumor size in recurrence HCC case [179]. Novel combinations of immune cell therapy were reported to be curative against PLC. Cocktail treatment composed of EGFR-CAR-T and CD133-CAR-T achieved a total of 13-month partial response in an advanced CCA patient [72]. Combination of DCs and CIKs was detected to recover the lung recurrence from liver undifferentiated embryonal sarcoma in one patient [180]. Further studies on the positive and negative impacts of combined cellular immunotherapy treatments are essential for clinical settings.
Obstacles and management for cellular immunotherapy in primary liver cancer
Cellular immune therapeutics have inaugurated a new generation of PLC therapies, however, obstacles such as cytokine release syndrome (CRS), loss of response (LOR), and organic adverse events still warrant further research for management [181, 182].
CRS, an overshooting inflammatory response triggered by iatrogenic or pathogenic causes, is the most notable adverse effects companied with immunotherapy and much important factor for evaluating the prognosis [183, 184]. From grade I to IV, CRS ranges a progressive severity of clinical presentations, and the severe multiple organ failure in grade IV can be life threaten [183]. T cell therapies, including CAR-T and other T cell engaged immunotherapies, are major iatrogenic causes for CRS in patients [184, 185]. Studies are sought to manage the CRS toxicity in CAR-T therapy and several approaches have been made to date. Cytokine inhibitors which aimed at blocking CRS in CAR-T treated patients were demonstrated to reduce the CRS toxicities in laboratory level. Itacitinib, a potent selective JAKI inhibitor, was detected to have potency on reducing CRS implicated cytokines in vitro and vivo, without suppression on antitumor efficiency. Further phase II clinical trial on validating the prophylaxis of itacitinib against CAR-T correlated CRS are initiated (NCT04071366) [186]. The application of tocilizumab, an antibody against IL6 receptor, also exhibited protective role in patients suffered from grade II to III CAR-T induced CRS [187]. Corticosteroids, continuous renal replacement therapy (CRRT), delivery optimization for immunotherapies and next generation CAR-T (with ON−/OFF-switch components or multiple antigen targetted gates) are also recommended for CRS management [185, 188,189,190].
Common categories for LOR, such as off-target effects and immune resistance, are blockages eager for solutions in cancer immunotherapy. Targeted at NK cell receptors (NKp46, CD16) and cancer cell antigens, NK cell engagers (NKCEs) reduced the off-target effects and revealed its integrated functions of both IgG antibodies multitude and tumor growth suppression, thus enhancing the antitumor efficiency of NK based immunotherapy [191]. Improved delivery strategies may also elevate precision and reduce the off-target effects for T cell based antitumor therapies [189]. Insufficient infiltration of CD8+T, aberrant expression of immune checkpoint molecules, and heterogeneity of individual genome, may all contribute to resistance in immunotherapy [192, 193]. Therefore, strategies to improve the management of therapeutic resistance need further exploration.
Organic adverse events vary from different strategies of immunotherapies and individual specificity, while similarly result in inferior prognosis and curative effects in patients. Complications such as neurotoxicity [194, 195], hepatotoxicity [196], infection [197, 198] and severe cutaneous adverse reactions [199] have been reported in engineered T cell therapy. Steroids are recommended for isolated immune effector cell associated neurotoxicity (ICANS) as first line therapy, while there are distinguished recommendations on managing grade I to IV stages for ICANS, followed the guideline of American society for transplantation and cellular therapy (ASTCT) [198]. For grade I ICANS, support treatment and monitoring are recommended, and for grade II to III, corticosteroids are indicated therapy, while for higher grade of ICANS, ICU guardian and airway protection are necessary in treatment [198, 200]. For hepatotoxicity induced by immune checkpoint inhibitors, corticosteroids are administered for grade II or higher hepatic lesion with symptoms [201]. Prophylaxes against infections, such as herpes simplex (HSV) and Pneumocystis jirovecii prophylaxis, are recommended after CAR-T therapy [198]. Further studies on the safety and adequate source of immune cells for PLC therapy are required [198, 202].
Conclusions
Immune cell based therapy is attractive for PLC treatment, especially the pioneering TCR-T/CAR-T approaches of adoptive cellular therapy. Mentioned as lymphoid or myeloid based cellular therapy, passive immunotherapies share the advantages on relatively unrestricted therapeutic patterns and have reached much progress in PLC therapy. On the other side, active cellular immunotherapy focuses on rebuilding the intrinsic immune microenvironment to exert its antineoplastic potency against PLC, which is superior in the risk reduction of uncertain triggered immune response while more in need of comprehensive considerations on the tumor immunogenicity and host’s immune status. Obstacles for cellular immunotherapy still remain and require preferable solutions when finally applied to PLC patients.
Availability of data and materials
Not applicable.
Abbreviations
- PLC:
-
Primary liver cancer
- HCC:
-
Hepatocellular carcinoma
- iCCA:
-
Intrahepatic cholangiocarcinoma
- TSAs/TAAs:
-
Tumor-specific/associated antigens
- APCs:
-
Antigen presenting cells
- DCs:
-
Dendritic cells
- GM-CSF:
-
Granulocyte macrophage colony stimulating factor
- IL-2:
-
Interleukin-2
- Treg:
-
Regulatory T cell
- Breg:
-
Regulatory B cell
- tTreg cells:
-
Thymus-derived naturally occurring Tregs
- pTreg cells:
-
Peripherally derived induced Tregs
- CTLA-4:
-
Cytotoxic T lymphocyte associated antigen-4
- TNF-α:
-
Tumor necrosis factor-α
- TLR-4:
-
Toll like receptor-4
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed cell death-ligand 1
- TGF-β:
-
Transforming growth factor β
- FOXP3:
-
Forkhead box protein P3
- GITRL:
-
Ligand to Tregs evoked glucocorticoid induced tumor necrosis factor receptor
- HGF:
-
Hepatocyte growth factor
- HMGB1:
-
High mobility group box 1
- MAPK:
-
Mitogen activated protein kinase
- TIM-1+ :
-
T cell immunoglobulin mucin domain-1 positive
- TCR:
-
T cell receptor
- CD4+ T:
-
CD3+CD4+CD8− αβTCR helper T cells
- CD8+ T:
-
CD3+CD4−CD8+ αβTCR cytotoxic T cells
- γδ T:
-
CD3+CD4−CD8+ αβTCR cytotoxic T cells
- EGFR:
-
Epidermal growth factor receptor
- TCR-T:
-
TCR engineered T cells
- CAR-T:
-
Chimeric antibody receptor engineered T Cells
- MHC:
-
Major histocompatibility complex
- AFP:
-
A-fetoprotein
- HLA:
-
Human leukocyte antigen
- PBMC:
-
Peripheral blood mononuclear cell
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- GPC3:
-
Glypican-3
- CXCR2:
-
C-X-C motif chemokine receptor 2
- PDX:
-
Patient derived xenograft
- NKG2D:
-
NK group 2 member D
- NKG2DL:
-
NKG2D ligands
- EGFRvIII:
-
Epidermal growth factor receptor variant III
- INF-γ:
-
Interferon-γ
- OS:
-
Overall survival
- PFS:
-
Progression free survival
- VEGF:
-
Vascular endothelial growth factor
- SDF-1:
-
Stromal cell derived factor 1
- EPC:
-
Endothelial progenitor cell
- PR:
-
Partial response
- mAb:
-
Monoclonal antibody
- NK cells:
-
Natural killer cells
- CXCL9:
-
C-X-C motif chemokine ligand-9
- DAP10:
-
DNAX-activating protein 10
- hIFN-α:
-
Human interferon-α
- MDSCs:
-
Myeloid derived suppressor cells
- TEMs:
-
TIE-2 expressing monocytes
- TANs:
-
Tumor associated neutrophils
- CIK:
-
DC-cytokine induced killer
- CRS:
-
Cytokine release syndrome
- LOR:
-
Loss of response
- CRRT:
-
Continuous renal replacement therapy
- NKCEs:
-
NK cell engagers
- ICANS:
-
Immune effector cell associated neurotoxicity
- ASTCT:
-
American society for transplantation and cellular therapy
- HSV:
-
Herpes simplex
- FGF:
-
Fibroblastic growth factor
- FGFR:
-
Fibroblastic growth factor receptor
- M-MDSC:
-
Monocytic MDSCs
- PMN-MDSCs:
-
Polymorphonuclear MDSCs
- TAM:
-
Tumor associated macrophages
- CCRK:
-
Cell cycle-related kinase
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
Dai W, Xu L, Yu X, Zhang G, Guo H, Liu H, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72(5):909–23.
Feng J, Li J, Wu L, Yu Q, Ji J, Wu J, et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39(1):1–19.
Wu L, Feng J, Li J, Yu Q, Ji J, Wu J, et al. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed Pharmacother. 2021;133:111036.
Yu Q, Wu L, Ji J, Feng J, Dai W, Li J, et al. Gut microbiota, peroxisome proliferator-activated receptors, and hepatocellular carcinoma. J Hepatocell Carcinoma. 2020;7:271–88.
Liu T, Li S, Wu L, Yu Q, Li J, Feng J, et al. Experimental study of hepatocellular carcinoma treatment by Shikonin through regulating PKM2. J Hepatocell Carcinoma. 2020;7:19–31.
Wu L, Li J, Liu T, Li S, Feng J, Yu Q, et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019;8(10):4806–20.
Feng J, Wu L, Ji J, Chen K, Yu Q, Zhang J, et al. PKM2 is the target of proanthocyanidin B2 during the inhibition of hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):1–15.
Feng J, Dai W, Mao Y, Wu L, Li J, Chen K, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):1–18.
Liu CY, Chen KF, Chen PJ. Treatment of liver Cancer. Cold Spring Harb Perspect Med. 2015;5(9):a021535.
Sia D, Villanueva A, Friedman SL, Llovet JM. Liver Cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152(4):745–61.
Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121(1):1–14.
Vergati M, Intrivici C, Huen NY, Schlom J, Tsang KY. Strategies for cancer vaccine development. J Biomed Biotechnol. 2010;2010:2–16.
Goldman B, DeFrancesco L. The cancer vaccine roller coaster. Nat Biotechnol. 2009;27(2):129–39.
Rojas-Sepulveda D, Tittarelli A, Gleisner MA, Avalos I, Pereda C, Gallegos I, et al. Tumor lysate-based vaccines: on the road to immunotherapy for gallbladder cancer. Cancer Immunol Immunother. 2018;67(12):1897–910.
Cao DY, Yang JY, Yue SQ, Tao KS, Song ZS, Wang DS, et al. Comparative analysis of DC fused with allogeneic hepatocellular carcinoma cell line HepG2 and autologous tumor cells as potential cancer vaccines against hepatocellular carcinoma. Cell Immunol. 2009;259(1):13–20.
Yang JY, Cao DY, Ma LY, Liu WC. Dendritic cells fused with allogeneic hepatocellular carcinoma cell line compared with fused autologous tumor cells as hepatocellular carcinoma vaccines. Hepatol Res. 2010;40(5):505–13.
Junking M, Grainok J, Thepmalee C, Wongkham S, Yenchitsomanus PT. Enhanced cytotoxic activity of effector T-cells against cholangiocarcinoma by dendritic cells pulsed with pooled mRNA. Tumour Biol. 2017;39(10):1010428317733367.
Rizell M, Sternby Eilard M, Andersson M, Andersson B, Karlsson-Parra A, Suenaert P. Phase 1 trial with the cell-based immune primer Ilixadencel, alone, and combined with Sorafenib, in advanced hepatocellular carcinoma. Front Oncol. 2019;9:19.
Peng BG, Liu SQ, Kuang M, He Q, Totsuka S, Huang L, et al. Autologous fixed tumor vaccine: a formulation with cytokine-microparticles for protective immunity against recurrence of human hepatocellular carcinoma. Jpn J Cancer Res. 2002;93(4):363–8.
Kuang M, Peng BG, Lu MD, Liang LJ, Huang JF, He Q, et al. Phase II randomized trial of autologous formalin-fixed tumor vaccine for postsurgical recurrence of hepatocellular carcinoma. Clin Cancer Res. 2004;10(5):1574–9.
Nemunaitis J, Barve M, Orr D, Kuhn J, Magee M, Lamont J, et al. Summary of bi-shRNA/GM-CSF augmented autologous tumor cell immunotherapy (FANG™) in advanced cancer of the liver. Oncology. 2014;87(1):21–9.
Sun TY, Yan W, Yang CM, Zhang LF, Tang HL, Chen Y, et al. Clinical research on dendritic cell vaccines to prevent postoperative recurrence and metastasis of liver cancer. Genet Mol Res. 2015;14(4):16222–32.
Iwashita Y, Tahara K, Goto S, Sasaki A, Kai S, Seike M, et al. A phase I study of autologous dendritic cell-based immunotherapy for patients with unresectable primary liver cancer. Cancer Immunol Immunother. 2003;52(3):155–61.
Panya A, Thepmalee C, Sawasdee N, Sujjitjoon J, Phanthaphol N, Junking M, et al. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol Immunother. 2018;67(10):1579–88.
Shimizu K, Kotera Y, Aruga A, Takeshita N, Takasaki K, Yamamoto M. Clinical utilization of postoperative dendritic cell vaccine plus activated T-cell transfer in patients with intrahepatic cholangiocarcinoma. J Hepatobil Pancreat Sci. 2012;19(2):171–8.
Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72(7):1853–64.
Trefzer U, Weingart G, Chen Y, Herberth G, Adrian K, Winter H, et al. Hybrid cell vaccination for cancer immune therapy: first clinical trial with metastatic melanoma. Int J Cancer. 2000;85(5):618–26.
Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, et al. Sorafenib for advanced and refractory Desmoid tumors. N Engl J Med. 2018;379(25):2417–28.
Lee WC, Wang HC, Hung CF, Huang PF, Lia CR, Chen MF. Vaccination of advanced hepatocellular carcinoma patients with tumor lysate-pulsed dendritic cells: a clinical trial. J Immunother. 2005;28(5):496–504.
Pedroza-Gonzalez A, Verhoef C, Ijzermans JN, Peppelenbosch MP, Kwekkeboom J, Verheij J, et al. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology. 2013;57(1):183–94.
Shao Y, Lo CM, Ling CC, Liu XB, Ng KT, Chu AC, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014;355(2):264–72.
Chen T, Song D, Min Z, Wang X, Gu Y, Wei B, et al. Perioperative dynamic alterations in peripheral regulatory T and B cells in patients with hepatocellular carcinoma. J Transl Med. 2012;10:14.
Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14(4):307–8.
Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–18.
Yi Y, He HW, Wang JX, Cai XY, Li YW, Zhou J, et al. The functional impairment of HCC-infiltrating gammadelta T cells, partially mediated by regulatory T cells in a TGFbeta- and IL-10-dependent manner. J Hepatol. 2013;58(5):977–83.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T Cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–56. e16.
Feng X, Li B, Ye H, Long D. Increased frequency of CD4+CD25high FoxP3+ regulatory T cells in patients with hepatocellular carcinoma. Arch Immunol Ther Exp. 2011;59(4):309–14.
Liu F, Liu W, Sanin DE, Jia G, Tian M, Wang H, et al. Heterogeneity of exhausted T cells in the tumor microenvironment is linked to patient survival following resection in hepatocellular carcinoma. Oncoimmunology. 2020;9(1):1746573.
Chen X, Du Y, Huang Z. CD4+CD25+ Treg derived from hepatocellular carcinoma mice inhibits tumor immunity. Immunol Lett. 2012;148(1):83–9.
Yang J, Zhang JX, Wang H, Wang GL, Hu QG, Zheng QC. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression via Treg requires TLR4 signaling. World J Gastroenterol. 2012;18(23):2938–47.
Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP+CTLA-4+Foxp3+ T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73(8):2435–44.
Huang Y, Wang FM, Wang T, Wang YJ, Zhu ZY, Gao YT, et al. Tumor-infiltrating FoxP3+ Tregs and CD8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion. 2012;86(4):329–37.
Yang XH, Yamagiwa S, Ichida T, Matsuda Y, Sugahara S, Watanabe H, et al. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol. 2006;45(2):254–62.
Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology. 2005;41(4):722–30.
Gao J, Ying Y, Wang J, Cui Y. Solanine inhibits immune escape mediated by Hepatoma Treg cells via the TGFbeta/Smad signaling pathway. Biomed Res Int. 2020;2020:9749631.
Li Q, Bao JM, Li XL, Zhang T, Shen XH. Inhibiting effect of Astragalus polysaccharides on the functions of CD4+CD25 highTreg cells in the tumor microenvironment of human hepatocellular carcinoma. Chin Med J. 2012;125(5):786–93.
Pedroza-Gonzalez A, Zhou G, Singh SP, Boor PP, Pan Q, Grunhagen D, et al. GITR engagement in combination with CTLA-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells ex vivo. Oncoimmunology. 2015;4(12):e1051297.
Sarvaria A, Madrigal JA, Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol. 2017;14(8):662–74.
Baba Y, Saito Y, Kotetsu Y. Heterogeneous subsets of B-lineage regulatory cells (Breg cells). Int Immunol. 2020;32(3):155–62.
Song S, Yuan P, Li P, Wu H, Lu J, Wei W. Dynamic analysis of tumor-associated immune cells in DEN-induced rat hepatocellular carcinoma. Int Immunopharmacol. 2014;22(2):392–9.
**ao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD-1hi identifies a novel regulatory B-cell population in human Hepatoma that promotes disease progression. Cancer Discov. 2016;6(5):546–59.
Ye L, Zhang Q, Cheng Y, Chen X, Wang G, Shi M, et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1(+) regulatory B cell expansion. J Immunother Cancer. 2018;6(1):145.
Ren Z, Peng H, Fu YX. PD-1 shapes B cells as evildoers in the tumor microenvironment. Cancer Discov. 2016;6(5):477–8.
Song SS, Yuan PF, Li PP, Wu HX, Ni WJ, Lu JT, et al. Protective effects of Total Glucosides of Paeony on N-nitrosodiethylamine-induced hepatocellular carcinoma in rats via Down-regulation of regulatory B cells. Immunol Investig. 2015;44(6):521–35.
Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202–13.
**a A, Zhang Y, Xu J, Yin T, Lu XJ. T cell dysfunction in Cancer immunity and immunotherapy. Front Immunol. 2019;10:1719.
Zheng BH, Ma JQ, Tian LY, Dong LQ, Song GH, Pan JM, et al. The distribution of immune cells within combined hepatocellular carcinoma and cholangiocarcinoma predicts clinical outcome. Clin Transl Med. 2020;10(1):45–56.
Huang CY, Wang H, Liao W, Han F, Li YQ, Chen SW, et al. Transforming growth factor β is a poor prognostic factor and inhibits the favorable prognostic value of CD8+ CTL in human hepatocellular carcinoma. J Immunother. 2017;40(5):175–86.
Lozano T, Chocarro S, Martin C, Lasarte-Cia A, Del Valle C, Gorraiz M, et al. Genetic modification of CD8+ T cells to express EGFR: potential application for adoptive T cell therapies. Front Immunol. 2019;10:2990.
Moreno-Cubero E, Larrubia JR. Specific CD8+ T cell response immunotherapy for hepatocellular carcinoma and viral hepatitis. World J Gastroenterol. 2016;22(28):6469–83.
Inada Y, Mizukoshi E, Seike T, Tamai T, Iida N, Kitahara M, et al. Characteristics of immune response to tumor-associated antigens and immune cell profile in patients with hepatocellular carcinoma. Hepatology. 2019;69(2):653–65.
Alnaggar M, Xu Y, Li J, He J, Chen J, Li M, et al. Allogenic Vgamma9Vdelta2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J Immunother Cancer. 2019;7(1):36.
Zhao L, Cao YJ. Engineered T cell therapy for Cancer in the clinic. Front Immunol. 2019;10:2250.
Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q, et al. Identification of alpha-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology. 2018;68(2):574–89.
Li Z, Gong H, Liu Q, Wu W, Cheng J, Mei Y, et al. Identification of an HLA-A*24:02-restricted alpha-fetoprotein signal peptide-derived antigen and its specific T-cell receptor for T-cell immunotherapy. Immunology. 2020;159(4):384–92.
Tan AT, Yang N, Lee Krishnamoorthy T, Oei V, Chua A, Zhao X, et al. Use of expression profiles of HBV-DNA integrated into genomes of Hepatocellular Carcinoma Cells to Select T Cells for Immunotherapy. Gastroenterology. 2019;156(6):1862–76. e9.
Qasim W, Brunetto M, Gehring AJ, Xue SA, Schurich A, Khakpoor A, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol. 2015;62(2):486–91.
Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived Xenografts of hepatocellular carcinoma. Front Immunol. 2016;7:690.
Dai H, Tong C, Shi D, Chen M, Guo Y, Chen D, et al. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: a single-arm, open-label, phase II trial. Oncoimmunology. 2020;9(1):1846926.
Feng KC, Guo YL, Liu Y, Dai HR, Wang Y, Lv HY, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10(1):1–11.
Luo X, Cui H, Cai L, Zhu W, Yang WC, Patrick M, et al. Selection of a clinical Lead TCR targeting alpha-fetoprotein-positive liver Cancer based on a balance of risk and benefit. Front Immunol. 2020;11:623.
Liu Q, Tian Y, Li Y, Zhang W, Cai W, Liu Y, et al. In vivo therapeutic effects of affinity-improved-TCR engineered T-cells on HBV-related hepatocellular carcinoma. J Immunother Cancer. 2020;8(2).
Spear TT, Callender GG, Roszkowski JJ, Moxley KM, Simms PE, Foley KC, et al. TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer Immunol Immunother. 2016;65(3):293–304.
Gehring AJ, Xue SA, Ho ZZ, Teoh D, Ruedl C, Chia A, et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J Hepatol. 2011;55(1):103–10.
Ma H, Chen S, He Y, Huang J, Xu Y, Wang C, et al. Redirecting T cells to glypican-3 with 28.41BB.Zeta and 28.Zeta-41BBL CARs for hepatocellular carcinoma treatment. Protein Cell. 2018;9(7):664–9.
Liu X, Gao F, Jiang L, Jia M, Ao L, Lu M, et al. 32A9, a novel human antibody for designing an immunotoxin and CAR-T cells against glypican-3 in hepatocellular carcinoma. J Transl Med. 2020;18(1):1–12.
Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-specific CAR T cells Coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. 2020;8(3):309–20.
Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020;50(5):712–24.
Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined antitumor effects of Sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol Ther. 2019;27(8):1483–94.
Liu X, Wen J, Yi H, Hou X, Yin Y, Ye G, et al. Split chimeric antigen receptor-modified T cells targeting glypican-3 suppress hepatocellular carcinoma growth with reduced cytokine release. Ther Adv Med Oncol. 2020;12:1758835920910347.
Sun B, Yang D, Dai H, Liu X, Jia R, Cui X, et al. Eradication of hepatocellular carcinoma by NKG2D-based CAR-T cells. Cancer Immunol Res. 2019;7(11):1813–23.
Liu H, Xu Y, **ang J, Long L, Green S, Yang Z, et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver Cancer. Clin Cancer Res. 2017;23(2):478–88.
Wang H, Ye X, Ju Y, Cai Z, Wang X, Du P, et al. Minicircle DNA-mediated CAR T cells targeting CD44 suppressed hepatocellular carcinoma both in vitro and in vivo. Onco Targets Ther. 2020;13:3703–16.
Ma Y, Chen Y, Yan L, Cao HX, Han SY, Cui JJ, et al. EGFRvIII-specific CAR-T cells produced by piggyBac transposon exhibit efficient growth suppression against hepatocellular carcinoma. Int J Med Sci. 2020;17(10):1406–14.
Wang Y, Liu J, Burrows PD, Wang JY. B cell development and maturation. Adv Exp Med Biol. 2020;1254:1–22.
Franchina DG, Grusdat M, Brenner D. B-cell metabolic remodeling and Cancer. Trends Cancer. 2018;4(2):138–50.
Abdelwahab FA, Hassanein KM, Hetta HF, Abdelmalek MO, Zahran AM, El-Badawy O. Impact of deranged B cell subsets distribution in the development of HCV-related cirrhosis and HCC in type two diabetes mellitus. Sci Rep. 2020;10(1):1–11.
Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology. 2018;67(5):1970–85.
Zhang Z, Ma L, Goswami S, Ma J, Zheng B, Duan M, et al. Landscape of infiltrating B cells and their clinical significance in human hepatocellular carcinoma. Oncoimmunology. 2019;8(4):e1571388.
Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66(2):342–51.
Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–43.
Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, et al. CD40-mediated immune cell activation enhances response to anti-PD1 in murine intrahepatic cholangiocarcinoma. J Hepatol. 2020;74:1145–54.
Medina-Echeverz J, Ma C, Duffy AG, Eggert T, Hawk N, Kleiner DE, et al. Systemic agonistic anti-CD40 treatment of tumor-bearing mice modulates hepatic myeloid-suppressive cells and causes immune-mediated liver damage. Cancer Immunol Res. 2015;3(5):557–66.
Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132(3):515–25.
Male V, Stegmann KA, Easom NJ, Maini MK. Natural killer cells in liver disease. Semin Liver Dis. 2017;37(3):198–209.
Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57(3):1107–16.
Sun H, Xu J, Huang Q, Huang M, Li K, Qu K, et al. Reduced CD160 expression contributes to impaired NK-cell function and poor clinical outcomes in patients with HCC. Cancer Res. 2018;78(23):6581–93.
Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. 2019;70(1):168–83.
Zhang QF, Yin WW, **a Y, Yi YY, He QF, Wang X, et al. Liver-infiltrating CD11b(−)CD27(−) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol. 2017;14(10):819–29.
Hodgins JJ, Khan ST, Park MM, Auer RC, Ardolino M. Killers 2.0: NK cell therapies at the forefront of cancer control. J Clin Invest. 2019;129(9):3499–510.
Kamiya T, Chang YH, Campana D. Expanded and activated natural killer cells for immunotherapy of hepatocellular carcinoma. Cancer Immunol Res. 2016;4(7):574–81.
Yu M, Luo H, Fan M, Wu X, Shi B, Di S, et al. Development of GPC3-specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol Ther. 2018;26(2):366–78.
Zhuang L, Fulton RJ, Rettman P, Sayan AE, Coad J, Al-Shamkhani A, et al. Activity of IL-12/15/18 primed natural killer cells against hepatocellular carcinoma. Hepatol Int. 2019;13(1):75–83.
Jiang W, Zhang C, Tian Z, Zhang J. hIFN-alpha gene modification augments human natural killer cell line anti-human hepatocellular carcinoma function. Gene Ther. 2013;20(11):1062–9.
Lin D, Lei L, Liu Y, Zhang Y, Hu B, Bao G, et al. Membrane IL1alpha inhibits the development of hepatocellular carcinoma via promoting T- and NK-cell activation. Cancer Res. 2016;76(11):3179–88.
Fukuda Y, Asaoka T, Eguchi H, Yokota Y, Kubo M, Kinoshita M, et al. Endogenous CXCL9 affects prognosis by regulating tumor-infiltrating natural killer cells in intrahepatic cholangiocarcinoma. Cancer Sci. 2020;111(2):323–33.
Su Z, Wang X, Zheng L, Lyu T, Figini M, Wang B, et al. MRI-guided interventional natural killer cell delivery for liver tumor treatment. Cancer Med. 2018;7(5):1860–9.
Lee MH, Liu KH, Thomas JL, Chen JR, Lin HY. Immunotherapy of Hepatocellular Carcinoma with Magnetic PD-1 Peptide-Imprinted Polymer Nanocomposite and Natural Killer Cells. Biomolecules. 2019;9(11):8–16.
Zhao Y, Zou W, Du J, Zhao Y. The origins and homeostasis of monocytes and tissue-resident macrophages in physiological situation. J Cell Physiol. 2018;233(10):6425–39.
Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. 2015;212(4):447–56.
Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–78.
Juric V, Ruffell B, Evason KJ, Hu J, Che L, Wang L, et al. Monocytes promote liver carcinogenesis in an oncogene-specific manner. J Hepatol. 2016;64(4):881–90.
Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9(8):793.
Meng YM, Liang J, Wu C, Xu J, Zeng DN, Yu XJ, et al. Monocytes/macrophages promote vascular CXCR4 expression via the ERK pathway in hepatocellular carcinoma. Oncoimmunology. 2018;7(3):e1408745.
Shen SL, Fu SJ, Huang XQ, Chen B, Kuang M, Li SQ, et al. Elevated preoperative peripheral blood monocyte count predicts poor prognosis for hepatocellular carcinoma after curative resection. BMC Cancer. 2014;14(1):1–9.
Lin ZX, Ruan DY, Li Y, Wu DH, Ma XK, Chen J, et al. Lymphocyte-to-monocyte ratio predicts survival of patients with hepatocellular carcinoma after curative resection. World J Gastroenterol. 2015;21(38):10898–906.
Ji J, Eggert T, Budhu A, Forgues M, Takai A, Dang H, et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology. 2015;62(2):481–95.
Hochst B, Schildberg FA, Sauerborn P, Gabel YA, Gevensleben H, Goltz D, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol. 2013;59(3):528–35.
Lee SWL, Adriani G, Ceccarello E, Pavesi A, Tan AT, Bertoletti A, et al. Characterizing the role of monocytes in T cell Cancer immunotherapy using a 3D microfluidic model. Front Immunol. 2018;9:416.
Chen DP, Ning WR, Li XF, Wei Y, Lao XM, Wang JC, et al. Peritumoral monocytes induce cancer cell autophagy to facilitate the progression of human hepatocellular carcinoma. Autophagy. 2018;14(8):1335–46.
Matsubara T, Kanto T, Kuroda S, Yoshio S, Higashitani K, Kakita N, et al. TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology. 2013;57(4):1416–25.
Shoji H, Yoshio S, Mano Y, Doi H, Sugiyama M, Osawa Y, et al. Pro-angiogenic TIE-2-expressing monocytes/TEMs as a biomarker of the effect of sorafenib in patients with advanced hepatocellular carcinoma. Int J Cancer. 2017;141(5):1011–7.
Dapas B, Grassi M, Grassi G. Can TIE-2 expressing monocytes represent a novel marker for hepatocellular carcinoma? Hepatobiliary Surg Nutr. 2014;3(4):175–8.
Kong L, Zhou Y, Bu H, Lv T, Shi Y, Yang J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J Exp Clin Cancer Res. 2016;35(1):1–11.
Keirsse J, Van Damme H, Geeraerts X, Beschin A, Raes G, Van Ginderachter JA. The role of hepatic macrophages in liver metastasis. Cell Immunol. 2018;330:202–15.
Trumper V, Wittig I, Heidler J, Richter F, Brune B, von Knethen A. Redox regulation of PPARgamma in polarized macrophages. PPAR Res. 2020;2020:8253831.
Wu L, Guo C, Wu J. Therapeutic potential of PPARgamma natural agonists in liver diseases. J Cell Mol Med. 2020;24(5):2736–48.
Yemchenko YO, Shynkevych VI, Ishcheikin KY, Kaidashev IP. PPAR-gamma agonist pioglitazone reduced CD68+ but not CD163+ macrophage dermal infiltration in obese psoriatic patients. PPAR Res. 2020;2020:4548012.
Zhang Y, Li JQ, Jiang ZZ, Li L, Wu Y, Zheng L. CD169 identifies an anti-tumour macrophage subpopulation in human hepatocellular carcinoma. J Pathol. 2016;239(2):231–41.
Kono H, Fujii H, Furuya S, Hara M, Hirayama K, Akazawa Y, et al. Macrophage colony-stimulating factor expressed in non-cancer tissues provides predictive powers for recurrence in hepatocellular carcinoma. World J Gastroenterol. 2016;22(39):8779–89.
Thanee M, Loilome W, Techasen A, Namwat N, Boonmars T, Pairojkul C, et al. Quantitative changes in tumor-associated M2 macrophages characterize cholangiocarcinoma and their association with metastasis. Asian Pac J Cancer Prev. 2015;16(7):3043–50.
Guerra AD, Yeung OWH, Qi X, Kao WJ, Man K. The anti-tumor effects of M1 macrophage-loaded poly (ethylene glycol) and gelatin-based hydrogels on hepatocellular carcinoma. Theranostics. 2017;7(15):3732–44.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–20.
Hetta HF, Zahran AM, Mansor SG, Abdel-Malek MO, Mekky MA, Abbas WA. Frequency and Implications of myeloid-derived suppressor cells and lymphocyte subsets in Egyptian patients with hepatitis C virus-related hepatocellular carcinoma. J Med Virol. 2019;91(7):1319–28.
Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. 2017;36(8):1090–101.
Liu M, Zhou J, Liu X, Feng Y, Yang W, Wu F, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut. 2020;69(2):365–79.
**e Y, Zhang Y, Wei X, Zhou C, Huang Y, Zhu X, et al. Jianpi Huayu decoction attenuates the immunosuppressive status of H22 hepatocellular carcinoma-bearing mice: by targeting myeloid-derived suppressor cells. Front Pharmacol. 2020;11:16.
Bassler K, Schulte-Schrep** J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The myeloid cell compartment-cell by cell. Annu Rev Immunol. 2019;37:269–93.
Geering B, Stoeckle C, Conus S, Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34(8):398–409.
Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130(10):5380–96.
Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. 2019;133(20):2159–67.
van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. 2018;68(4):1347–60.
Margetts J, Ogle LF, Chan SL, Chan AWH, Chan KCA, Jamieson D, et al. Neutrophils: driving progression and poor prognosis in hepatocellular carcinoma? Br J Cancer. 2018;118(2):248–57.
Wong L, Bozhilov K, Hernandez B, Kwee S, Chan O, Ellis L, et al. Underlying liver disease and advanced stage liver cancer are associated with elevated neutrophil-lymphocyte ratio. Clin Mol Hepatol. 2019;25(3):305–16.
Okamura Y, Sugiura T, Ito T, Yamamoto Y, Ashida R, Mori K, et al. Neutrophil to lymphocyte ratio as an indicator of the malignant behaviour of hepatocellular carcinoma. Br J Surg. 2016;103(7):891–8.
Zhou SL, Yin D, Hu ZQ, Luo CB, Zhou ZJ, **n HY, et al. A positive feedback loop between Cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology. 2019;70(4):1214–30.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150(7):1646–58. e17.
Nan J, **ng YF, Hu B, Tang JX, Dong HM, He YM, et al. Endoplasmic reticulum stress induced LOX-1(+ ) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154(1):144–55.
Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):931–44.
Alnaggar M, Lin M, Mesmar A, Liang S, Qaid A, Xu K, et al. Allogenic natural killer cell immunotherapy combined with irreversible electroporation for stage IV hepatocellular carcinoma: survival outcome. Cell Physiol Biochem. 2018;48(5):1882–93.
He W, Huang Z, Zhou S, Huang L, Wang B, Zhu L, et al. The effect of DC+CIK combined therapy on rat liver cancer model and its modulatory effect on immune functions. Eur Rev Med Pharmacol Sci. 2018;22(3):778–85.
Niu LZ, Li JL, Zeng JY, Mu F, Liao MT, Yao F, et al. Combination treatment with comprehensive cryoablation and immunotherapy in metastatic hepatocellular cancer. World J Gastroenterol. 2013;19(22):3473–80.
Zhou P, Liang P, Dong B, Yu X, Han Z, Xu Y. Phase clinical study of combination therapy with microwave ablation and cellular immunotherapy in hepatocellular carcinoma. Cancer Biol Ther. 2011;11(5):450–6.
Behm B, Di Fazio P, Michl P, Neureiter D, Kemmerling R, Hahn EG, et al. Additive antitumour response to the rabbit VX2 hepatoma by combined radio frequency ablation and toll like receptor 9 stimulation. Gut. 2016;65(1):134–43.
Hosseinzadeh F, Verdi J, Ai J, Hajighasemlou S, Seyhoun I, Parvizpour F, et al. Combinational immune-cell therapy of natural killer cells and sorafenib for advanced hepatocellular carcinoma: a review. Cancer Cell Int. 2018;18(1):1–12.
Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34(2):280–300.
Raja A, Park I, Haq F, Ahn SM. FGF19-FGFR4 Signaling in Hepatocellular Carcinoma. Cells. 2019;8(6):158–162.
Lu X, Chen H, Patterson AV, Smaill JB, Ding K. Fibroblast growth factor receptor 4 (FGFR4) selective inhibitors as hepatocellular carcinoma therapy: advances and prospects. J Med Chem. 2019;62(6):2905–15.
Wang Y, Liu D, Zhang T, **a L. FGF/FGFR signaling in Hepatocellular Carcinoma: From Carcinogenesis to recent therapeutic intervention. Cancers. 2021;13(6):158–162.
Jo JC, Choi EK, Shin JS, Moon JH, Hong SW, Lee HR, et al. Targeting FGFR pathway in human hepatocellular carcinoma: expressing pFGFR and pMET for antitumor activity. Mol Cancer Ther. 2015;14(11):2613–22.
Huynh H, Lee LY, Goh KY, Ong R, Hao HX, Huang A, et al. Infigratinib mediates vascular normalization, impairs metastasis, and improves chemotherapy in hepatocellular carcinoma. Hepatology. 2019;69(3):943–58.
Le TBU, Vu TC, Ho RZW, Prawira A, Wang L, Goh BC, et al. Bevacizumab augments the antitumor efficacy of infigratinib in Hepatocellular Carcinoma. Int J Mol Sci. 2020;21(24).
Chun YS, Javle M. Systemic and adjuvant therapies for intrahepatic Cholangiocarcinoma. Cancer Control. 2017;24(3):1073274817729241.
Peng X, Hou P, Chen Y, Dai Y, Ji Y, Shen Y, et al. Preclinical evaluation of 3D185, a novel potent inhibitor of FGFR1/2/3 and CSF-1R, in FGFR-dependent and macrophage-dominant cancer models. J Exp Clin Cancer Res. 2019;38(1):372.
Wang N, Li JY, Li S, Guo XC, Wu T, Wang WF, et al. Fibroblast growth factor 21 regulates foam cells formation and inflammatory response in ox-LDL-induced THP-1 macrophages. Biomed Pharmacother. 2018;108:1825–34.
Han X, Li L, Yang J, King G, **ao Z, Quarles LD. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS Lett. 2016;590(1):53–67.
Wang N, Li JY, Zhao TT, Li SM, Shen CB, Li DS, et al. FGF-21 plays a crucial role in the glucose uptake of activated monocytes. Inflammation. 2018;41(1):73–80.
Haddad LE, Khzam LB, Hajjar F, Merhi Y, Sirois MG. Characterization of FGF receptor expression in human neutrophils and their contribution to chemotaxis. Am J Physiol Cell Physiol. 2011;301(5):C1036–45.
Somasundaram R, Zhang G, Fukunaga-Kalabis M, Perego M, Krepler C, Xu X, et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat Commun. 2017;8(1):607.
Hiraoka A, Kumada T, Fukunishi S, Atsukawa M, Hirooka M, Tsuji K, et al. Post-progression treatment eligibility of Unresectable hepatocellular carcinoma patients treated with Lenvatinib. Liver Cancer. 2020;9(1):73–83.
Alsina A, Kudo M, Vogel A, Cheng AL, Tak WY, Ryoo BY, et al. Effects of subsequent systemic anticancer medication following first-line Lenvatinib: a post hoc responder analysis from the phase 3 REFLECT study in Unresectable hepatocellular carcinoma. Liver Cancer. 2020;9(1):93–104.
Chen WX, Li GX, Hu ZN, Zhu P, Zhang BX, Ding ZY. Significant response to anti-PD-1 based immunotherapy plus lenvatinib for recurrent intrahepatic cholangiocarcinoma with bone metastasis: a case report and literature review. Medicine (Baltimore). 2019;98(45):e17832.
Kato Y, Tabata K, Kimura T, Yachie-Kinoshita A, Ozawa Y, Yamada K, et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS One. 2019;14(2):e0212513.
Kimura T, Kato Y, Ozawa Y, Kodama K, Ito J, Ichikawa K, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018;109(12):3993–4002.
Deng H, Kan A, Lyu N, Mu L, Han Y, Liu L, et al. Dual vascular endothelial growth factor receptor and fibroblast growth factor receptor inhibition elicits antitumor immunity and enhances programmed cell Death-1 checkpoint blockade in hepatocellular carcinoma. Liver Cancer. 2020;9(3):338–57.
Zhang J, Wu N, Lian Z, Feng H, Jiang Q, Chen X, et al. The combined antitumor effects of (125) I radioactive particle implantation and cytokine-induced killer cell therapy on Xenograft hepatocellular carcinoma in a mouse model. Technol Cancer Res Treat. 2017;16(6):1083–91.
**e S, Wu Z, Zhou L, Liang Y, Wang X, Niu L, et al. Iodine-125 seed implantation and allogenic natural killer cell immunotherapy for hepatocellular carcinoma after liver transplantation: a case report. Onco Targets Ther. 2018;11:7345–52.
**e S, Wu X, Zhang G, Xu K, Bian X, Zhang S, et al. Remarkable regression of a lung recurrence from an undifferentiated embryonal sarcoma of the liver treated with a DC vaccine combined with immune cells: a case report. Cell Immunol. 2014;290(2):185–9.
Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104.
DeFrancesco L. CAR-T cell therapy seeks strategies to harness cytokine storm. Nat Biotechnol. 2014;32(7):604.
Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):1–14.
Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383(23):2255–73.
Liu D, Zhao J. Cytokine release syndrome: grading, modeling, and new therapy. J Hematol Oncol. 2018;11(1):1–7.
Huarte E, O'Connor RS, Peel MT, Nunez-Cruz S, Leferovich J, Juvekar A, et al. Itacitinib (INCB039110), a JAK1 inhibitor, reduces cytokines associated with cytokine release syndrome induced by CAR T-cell therapy. Clin Cancer Res. 2020;26(23):6299–309.
Chen H, Wang F, Zhang P, Zhang Y, Chen Y, Fan X, et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front Med. 2019;13(5):610–7.
Constantinescu C, Pasca S, Tat T, Teodorescu P, Vlad C, Iluta S, et al. Continuous renal replacement therapy in cytokine release syndrome following immunotherapy or cellular therapies? J Immunother Cancer. 2020;8(1):185, 187–189.
Zhao Z, Zheng L, Chen W, Weng W, Song J, Ji J. Delivery strategies of cancer immunotherapy: recent advances and future perspectives. J Hematol Oncol. 2019;12(1):1–14.
Andrea AE, Chiron A, Bessoles S, Hacein-Bey-Abina S. Engineering next-generation CAR-T cells for better toxicity management. Int J Mol Sci. 2020;21(22):8620.
Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177(7):1701–13.e16.
Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9(379):191, 192.
Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. beta-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in Hepatocellular Carcinoma. Cancer Discov. 2019;9(8):1124–41.
Karschnia P, Jordan JT, Forst DA, Arrillaga-Romany IC, Batchelor TT, Baehring JM, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. 2019;133(20):2212–21.
Chou CK, Turtle CJ. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant. 2019;54(Suppl 2):780–4.
Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21(14):3149–59.
Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018;131(1):121–30.
Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Muller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48.
Pan RY, Chu MT, Wang CW, Lee YS, Lemonnier F, Michels AW, et al. Identification of drug-specific public TCR driving severe cutaneous adverse reactions. Nat Commun. 2019;10(1):3569.
Azoulay E, Shimabukuro-Vornhagen A, Darmon M, von Bergwelt-Baildon M. Critical Care Management of Chimeric Antigen Receptor T Cell-related Toxicity. Be aware and prepared. Am J Respir Crit Care Med. 2019;200(1):20–3.
Liu YH, Zang XY, Wang JC, Huang SS, Xu J, Zhang P. Diagnosis and Management of Immune Related Adverse Events (irAEs) in Cancer immunotherapy. Biomed Pharmacother. 2019;120:109437.
Cancelas JA. Granulocyte transfusion: questions remain. Blood. 2015;126(18):2082–3.
Acknowledgements
We give thanks to the Putuo People’s Hospital, Shanghai Tenth People’s Hospital and Tongji University for support.
Funding
This work was supported by the following grants: National Natural Science Foundation of China (No. 82002539), Yangfan Project of Shanghai Science and Technology Commission (No. 20YF1443300), Natural Science Foundation of Shanghai (No. 19ZR1447700), Health System Innovation Project of Shanghai Putuo Science and Technology Commission (No. PTKWWS201801, No. PTKWWS201903), WBN Hepatology Research Fund of China Hepatitis Prevention and Treatment Foundation (No. CFHPC2019031).
Author information
Authors and Affiliations
Contributions
YY Zheng, Y Li, CY Guo, YQ Zhou and JY Wu preliminarily constructed the conceptual framework. YY Zheng and Y Li originally wrote the draft manuscript. J Feng, JJ Li, J Ji, LW Wu, Q Yu and WQ Dai reviewed and edited the writing. YY Zheng visualized the tables and figures in the work. CY Guo supervised the work. YY Zheng and Y Li contributed equally to this manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, Y., Li, Y., Feng, J. et al. Cellular based immunotherapy for primary liver cancer. J Exp Clin Cancer Res 40, 250 (2021). https://doi.org/10.1186/s13046-021-02030-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02030-5