
Abstract
Background
Clinical studies on progressive familial intrahepatic cholestasis (PFIC) type 5 caused by mutations in NR1H4 are limited.
Methods
New patients with biallelic NR1H4 variants from our center and all patients from literature were retrospectively analyzed.
Results
Three new patients were identified to be carrying five new variants. Liver phenotypes of our patients manifests as low-γ-glutamyl transferase cholestasis, liver failure and related complications. One patient underwent liver transplantation (LT) and survived, and two other patients died without LT. Nine other patients were collected through literature review. Twelve out of 13 patients showed neonatal jaundice, with the median age of onset being 7 days after birth. Reported clinical manifestations included cholestasis (13/13, 100%), elevated AFP (11/11, 100%), coagulopathy (11/11, 100%), hypoglycemia (9/13, 69%), failure to thrive (8/13, 62%), splenomegaly (7/13, 54%), hyperammonemia (7/13, 54%), and hepatomegaly (6/13, 46%). Six of 13 patients received LT at a median age of 6.2 months, and only one patient died of acute infection at one year after LT. Other 7 patients had no LT and died with a median age of 5 months (range 1.2-8). There were 8 patients with homozygous genotype and 5 patients with compound heterozygous genotype. In total, 13 different variants were detected, and 5 out of 12 single or multiple nucleotides variants were located in exon 5.
Conclusions
We identified three newly-diagnosed patients and five novel mutations. NR1H4-related PFIC typically cause progressive disease and early death. LT may be the only lifesaving therapy leading to cure.
Similar content being viewed by others


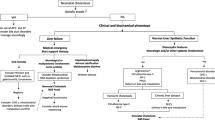
Background
Progressive familial intrahepatic cholestasis (PFIC) is an ever-growing group of autosomal recessive liver disorders caused by defects in genes associated with bile secretion, bile salt and lipid transporters and regulators [1, 2]. Disease-causing genes of PFIC were gradually revealed, including ATP8B1, ABCB11, ABCB4, TJP2, NR1H4, and MYO5B (named PFIC1 to 6) [3,4,5,6]. NR1H4 gene (OMIM *603,826), located on 12q23.1, encodes the farnesoid X receptor (FXR), a bile acid (BA)-activated transcription factor, and plays an essential role in BA homeostasis [7]. Biallelic pathogenic variants in NR1H4 were first identified in low-γ-glutamyl transferase (GGT) cholestasis patients and termed as PFIC5 in 2016 4. So far, only 10 patients with PFIC5 from six unrelated families have been reported, usually presenting as rapidly progressive liver failure, vitamin K independent coagulopathy, high alpha-fetoprotein (AFP) and ultimately required a liver transplant (LT) to save lives [4, 8,9,10]. To assess phenotypic spectrum and clinical outcomes in NR1H4-related PFIC, we studied our patients in detail and reviewed previously reported patients in the literature.
Methods
Subjects
Our patients were all Chinese children referred to the Center for Pediatric Liver Disease, Children’s Hospital of Fudan University from February 2016 to March 2023. Genetic testing, either whole-exome sequencing or liver panel sequencing, was performed in patients after excluding other causes of liver diseases (including infection, drug exposure, autoimmune hepatitis, and biliary atresia) [11]. Cytomegalovirus infection was not excluded due to its high prevalence in Chinese infants. When other known inherited liver disorders were excluded, patients with two or biallelic NR1H4 pathogenic/likely pathogenic/uncertain significance (P/LP/US) variants were collected. P/LP/US variants were classified according to the American College of Medical Genetics (ACMG)/Association for Molecular Pathology (AMP) criteria [12].
Genetic analyses and in silico assessment of NR1H4 variants
We confirmed these variants by Sanger sequencing and confirmed parental origins when available. Assessment of variant pathogenicity were performed with seven in silico tools including MutationTaster (http://www.mutationtaster.org/), Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org), Rare Exome Variant Ensemble Learner (REVEL, https://labworm.com/tool/revel), MutPred (http://mutpred.mutdb.org/), and Protein Variation Effect Analyzer (PROVEAN, http://provean.jcvi.org/index.php). Two programs, SpliceAI (https://spliceailookup.broadinstitute.org/) and varSEAK (https://varseak.bio/), were used to evaluate the effect of variants on mRNA splicing. Default settings were used for all in silico tools.
Literature review
A comprehensive literature review of the current literature was performed on March 2023 by searching PubMed (https://pubmed.ncbi.nlm.nih.gov/), CNKI (https://www.cnki.net/), and Wan fang (https://www.wanfangdata.com.cn/) databases using the keywords “NR1H4 variants, progressive familial intrahepatic cholestasis 5, and PFIC5”.
Results
Identification of biallelic NR1H4 variants in 3 new patients and in silico assessment
We identified 5 unique NR1H4 variants from 3 unrelated Chinese patients with low GGT intrahepatic cholestasis from our cohort. Among these, there were 3 missense variants, one nonsense variant, and one canonical splicing variant. All variants have not been previously reported in medical literatures (See Table 1). Three out of 5 variants were absent in gnomAD (c.505T > A/p. (Cys169Ser), c.1235T > C/p. (Leu412Pro), and c.1066 + 1G > A/p.?). The other two were present in gnomAD, with a population frequency of 0/1/251,458 (number of homozygotes/allele count/allele number) for the c.688 C > T/p. (Arg230Ter) variant and 0/1/31,410 (number of homozygotes/allele count/allele number) for the c.527G > A/p. (Arg176Gln) variant. MutationTaster predicted that the nonsense variant (c.688 C > T/p. (Arg230Ter)) may lead to nonsense-mediated mRNA decay (NMD). SpliceAI and varSEAK predicted that the canonical splicing variant (c.1066 + 1G > A/p.?) lead to loss of donor splice site, most likely leading to protein truncation. Three missense variants were predicted to be pathogenic by five pathogenicity prediction tools, and have no effect on pre-mRNA splicing. According to the ACMG/AMP criteria, all of the variants were classified as P/LP/US (Table 1).
Clinical features and outcome of 3 new patients with NR1H4-related cholestasis
All 3 male patients born at full-term with normal weight to non-consanguineous parents following uneventful pregnancies. The first child in family II died of unexplained liver disorders at the age of 7 months, whereas the other families did not have positive family history. All of them presented with jaundice during the first few days after birth. Normal growth and development were observed in patient 1(P1), and failure-to-thrive occurred in patients 2 (P2) and 3 (P3). Our patients have similar clinical features resembling previously reported patients, including low-GGT cholestasis, rapidly progressive liver failure/decompensated cirrhosis, vitamin K independent coagulopathy, and markedly elevated AFP levels (Table 2). Initial, pre-transplantation, or pre-death laboratory testing results were shown in Table 3. In addition, our patients had recurrent severe pneumonia, splenomegaly, and elevated urinary microalbumin. Two patients (P1 and P3) had hepatomegaly and hydrocele.
The patient P1 suffered massive ascites from decompensated cirrhosis prior to liver transplantation (LT). He had a positive plasma Epstein-Barr virus DNA from 2 months post-LT until the last visit. He also underwent a single surgery due to small bowel obstruction and right-sided diaphragmatic hernia one year after LT. The patient P3 presented with hyperammonemia due to acute liver failure (ALF) and his magnetic resonance of the brain showed minor abnormalities such as widened extracerebral space and cavum septum pellucidum.
Only one patient (P1) was treated by LT at the age of 4 months, and the other two patients died of infection at 3 months of age. Up to the latest assessment, P1 had normal liver function at one-year post-transplant.
Clinical and genetic characterization of 13 NR1H4-related PFIC patients
A total of 13 patients from 10 unrelated families were collected, with 3 new and 10 reported patients (Supplementary Table 1) [4, 8,9,10]. Eight were males, 4 were females, and one patient’s gender was unknown. All patients were delivered at full term without maternal or fetal complications. The age of onset ranged from the neonatal period to 17 months, with the median age of onset being 7 days after birth. Twelve out of 13 patients showed neonatal jaundice in the early neonatal period (7 patients, 0–6 days) and late neonatal period (5 patients, 7–28 days), respectively. Only one patient was admitted to hospital due to jaundice and abdominal distention at the age of 17 months. Reported clinical manifestations include: cholestasis (13/13, 100%), persistently elevated AFP (11/11, 100%), coagulopathy (11/11, 100%), hypoglycemia (9/13, 69%), splenomegaly (7/13, 54%), hyperammonemia (7/13, 54%), failure to thrive (8/13, 62%), and hepatomegaly (6/13, 46%). Six of 13 patients received LT at a median age of 6.2 months (range 2–20). Only one patient died of acute infection at one year after LT. Five out of 6 patients with LT are still alive, with a median age of 6 years (range 1.3–10). Other 7 patients without LT died at a median age of 5 months (range 1.2-8). The causes of death included ALF, multiple organ dysfunction syndrome (MODS), sepsis, and others (Supplementary Table 1).
Eight patients had homozygous genotype and 5 patients had compound heterozygous genotype. In total, 13 different variants were detected, including 4 nonsense variants, 4 missense variants, 2 frameshift variants, one splice site variant, one in-frame insertion variant, and one large DNA fragment deletion variant. Overall, there were five variants (41.6%) in exon 5, two variants (16.7%) in exon 4, one variant (8.3%) in exon 6, one variant (8.3%) in exon 8, one variant (8.3%) in exon 9, one variant (8.3%) in intron 9 and one variant (8.3%) in exon 11 (Figure 1).

Schematic presentation of the farnesoid X receptor (FXR) protein and locations of 12 unique single or multiple nucleotides variants identified in our cohort and literature. The exon 5 was depicted in red, and other exons of the NR1H4 gene are depicted in blue; FXR protein domains are shown in green
Liver pathological characteristics in NR1H4-related PFIC patients
Liver pathology was documented in 7 of 13 NR1H4-related PFIC [8, 9]. Characteristic pathological features included cholestasis, steatosis, micro-nodular cirrhosis, hepatocellular ballooning, fibrous tissue proliferation, fibrosis, inflammatory cell infiltration, and proliferation of bile ducts. Immunohistochemical stainings of both bile salt export pump (BESP) and FXR proteins were absent, and the multi-drug resistance protein 3 (MDR3) expression was decreased or normal in all 7 patients.
Discussion
It was first discovered 7 years ago that the NR1H4 was responsible for PFIC [4]. Previous studies showed that the NR1H4-associated PFIC had early-onset and rapid disease progression with high mortality [4, 8]. As only few patients have been reported, current understanding of PFIC5 caused by NR1H4 defect is limited. Therefore, we performed a retrospective analysis to obtain a better understanding of clinical phenotype and outcomes of PFIC5 caused by NR1H4 in our center and literature.
The FXR, encoded by NR1H4 gene, as the master regulator of BA homeostasis, regulates BA homeostasis, biliary BA secretion, and intestinal re-absorption [13,14,15]. Compared with other PFIC patients, the PFIC5 patients caused by NR1H4 defect had significantly worse prognosis due to more rapid progression [8, 16, 17]. All patients without LT died and survival with native liver has not been observed. Three new patients in our cohort exhibited similar clinical characteristics as published case [7, 8]. The liver phenotypes of all reported patients were extremely similar and presented as low GGT neonatal cholestasis with rapid progression to ALF (with/without related complications such as hypoglycemia, hyperammonemia, coagulopathy, hepatosplenomegaly, hydrothorax, and ascites) [8, 9].
While NR1H4 gene is predominantly expressed in liver and intestine, it also presents in kidney, spleen, heart, gallbladder, pancreas, adrenal glands, bone marrow, and other tissues [18,19,20,21]. We further summarized the extra-hepatic phenotypes of 13 patients. Of those, the failure to thrive was the most common finding. Other extrahepatic manifestations were also described, such as atrial septal defect, butterfly vertebra, decreased bone mineral density, intestinal obstruction, diaphragmatic hernia, inguinal hernia, and iris coloboma. Although NR1H4 is highly expressed in the intestine, recurrent or severe diarrhea has not been observed in patients with NR1H4-related disorder [8,9,10].
Notably, pharmacological therapy is typically not effective for NR1H4 disease [8]. Those patients without LT died in early infancy, and the common causes of death included ALF, MODS, and severe infection [8]. Therefore, LT may be the only curative option. Fortunately, 5 out of 6 patients are still alive after LT without serious postoperative complications, and with good clinical outcome during the follow-up. Only one patient died of acute infections one month after the transplant [9], this suggests that minimizing the risk of infection is the key to reduce morbidity and mortality associated with LT for PFIC5 patients [22, 23].
All enrolled patients had poor prognosis with native liver. So, we were not able to analyze the relationship between genotypes and phenotype. However, we observed that 41.6% of all reported variants are located on the exon 5 of NR1H4 gene. Exon5 encodes a highly conserved DNA binding domain of FXR by binding to specific DNA sequences called hormone response elements, thereby possibly regulating other gene expression [24, 25]. More cases and further studies are needed to confirm whether exon 5 is a susceptible or hotspot region for NR1H4 gene mutation.
Conclusions
NR1H4-related PFIC is characterized by severe neonatal cholestasis, rapid progression to liver failure, and early death. LT might be the only lifesaving therapy that can lead to cure. At present, no severe complications of LT related to NR1H4 gene were observed, but long-term outcome of LT still needs to be validated in more patients.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- AMP:
-
Association for Molecular Pathology
- ACMG:
-
American College of Medical Genetics
- ALF:
-
Acute liver failure
- AFP:
-
High alpha-fetoprotein
- BA:
-
Bile acid
- BESP:
-
Bile salt export pump
- FXR:
-
Farnesoid X receptor
- GGT:
-
Glutamyl transferase
- LP:
-
Likely pathogenic
- LT:
-
Liver transplantation
- MDR3:
-
Multi-drug resistance protein 3
- NMD:
-
Nonsense-mediated mRNA decay
- P:
-
Pathogenic
- PFIC:
-
Progressive familial intrahepatic cholestasis
- US:
-
Uncertain significant
References
Sticova E, Jirsa M, Pawlowska J. New insights in genetic cholestasis: from molecular mechanisms to clinical implications. Can J Gastroenterol Hepatol. 2018; 2018:2313675.
Amirneni S, Haep N, Gad MA, Soto-Gutierrez A, Squires JE, Florentino RM. Molecular overview of progressive familial intrahepatic cholestasis. World J Gastroenterol. 2020;26:7470–84.
Vitale G, Gitto S, Vukotic R, Raimondi F, Andreone P. Familial intrahepatic cholestasis: new and wide perspectives. Dig Liver Dis. 2019;51:922–33.
Gomez-Ospina N, Potter CJ, **ao R, Manickam K, Kim MS, Kim KH, et al. Mutations in the nuclear bile acid receptor fxr cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:10713.
Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, et al. Mutations in tjp2 cause progressive cholestatic liver disease. Nat Genet. 2014;46:326–8.
Qiu YL, Gong JY, Feng JY, Wang RX, Han J, Liu T, et al. Defects in myosin vb are associated with a spectrum of previously undiagnosed low gamma-glutamyltransferase cholestasis. Hepatology. 2017;65:1655–69.
Cariello M, Piccinin E, Garcia-Irigoyen O, Sabba C, Moschetta A. Nuclear receptor fxr, bile acids and liver damage: introducing the progressive familial intrahepatic cholestasis with fxr mutations. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1308–18.
Himes RW, Mojarrad M, Eslahi A, Finegold MJ, Maroofian R, Moore DD. Nr1h4-related progressive familial intrahepatic cholestasis 5: further evidence for rapidly progressive liver failure. J Pediatr Gastroenterol Nutr. 2020;70:e111–3.
Czubkowski P, Thompson RJ, Jankowska I, Knisely AS, Finegold M, Parsons P, et al. Progressive familial intrahepatic cholestasis - farnesoid x receptor deficiency due to nr1h4 mutation: a case report. World J Clin Cases. 2021;9:3631–6.
Chen HL, Li HY, Wu JF, Wu SH, Chen HL, Yang YH, et al. Panel-based next-generation sequencing for the diagnosis of cholestatic genetic liver diseases: clinical utility and challenges. J Pediatr. 2019;205:153–9.
Yang L, Kong Y, Dong X, Hu L, Lin Y, Chen X, et al. Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet Med. 2019;21:564–71.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–24.
Chiang J, Ferrell JM. Discovery of farnesoid x receptor and its role in bile acid metabolism. Mol Cell Endocrinol. 2022;548:111618.
Rausch M, Samodelov SL, Visentin M, Kullak-Ublick GA. The farnesoid x receptor as a master regulator of hepatotoxicity. Int J Mol Sci 2022;23.
Keitel V, Droge C, Haussinger D. Targeting fxr in cholestasis. Handb Exp Pharmacol. 2019;256:299–324.
Hassan S, Hertel P. Overview of progressive familial intrahepatic cholestasis. Clin Liver Dis. 2022;26:371–90.
Pfister ED, Droge C, Liebe R, Stalke A, Buhl N, Ballauff A, et al. Extrahepatic manifestations of progressive familial intrahepatic cholestasis syndromes: presentation of a case series and literature review. Liver Int. 2022;42:1084–96.
Yan N, Yan T, **a Y, Hao H, Wang G, Gonzalez FJ. The pathophysiological function of non-gastrointestinal farnesoid x receptor. Pharmacol Ther. 2021;226:107867.
Fu T, Li Y, Oh TG, Cayabyab F, He N, Tang Q, et al. Fxr mediates ilc-intrinsic responses to intestinal inflammation. Proc Natl Acad Sci U S A. 2022;119:e2081926177.
Xu S, Jia P, Fang Y, ** J, Sun Z, Zhou W, et al. Nuclear farnesoid x receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 2022;101:987–1002.
Zheng Y, Sun W, Wang Z, Liu J, Shan C, He C et al. Activation of pancreatic acinar fxr protects against pancreatitis via osgin1-mediated restoration of efficient autophagy. Research (Wash D C). 2022; 2022:9784081.
Smith SK, Miloh T. Pediatric liver transplantation. Clin Liver Dis. 2022;26:521–35.
Selimoglu MA, Kaya S, Gungor S, Varol FI, Gozukara-Bag HG, Yilmaz S. Infection risk after paediatric liver transplantation. Turk J Pediatr. 2020;62:46–52.
Jiang L, Zhang H, **ao D, Wei H, Chen Y. Farnesoid x receptor (fxr): structures and ligands. Comput Struct Biotechnol J. 2021;19:2148–59.
Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD, Edwards PA. Identification of the dna binding specificity and potential target genes for the farnesoid x-activated receptor. J Biol Chem. 2000;275:10638–47.
Acknowledgements
The authors thank the patients’ families for their participation.
Funding
This study was supported by grants from Key Development Program of Children’s Hospital of Fudan University (EK2022ZX05).
Author information
Authors and Affiliations
Contributions
XXB designed and supervised the study, and involved in the draft and revision of the manuscript; LZD collected data and analyzed relevant information; LZD wrote the manuscript; LYC and ZJ provided clinical information for the work; WJS clinically managed patients. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval consent to participate
This study was approved by the ethics committees of Children’s Hospital of Fudan University, and complied with the guidelines of the 1975 Declaration of Helsinki (2020 − 402). Informed consent was waived the due to its retrospective nature.
Consent for publication
Consents for publication were obtained.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, ZD., Li, YC., **g-Zhao et al. NR1H4 disease: rapidly progressing neonatal intrahepatic cholestasis and early death. Orphanet J Rare Dis 19, 171 (2024). https://doi.org/10.1186/s13023-024-03166-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-024-03166-1