
Abstract
Background
Plant growth devices, for example, rhizoponics, rhizoboxes, and ecosystem fabrication (EcoFAB), have been developed to facilitate studies of plant root morphology and plant-microbe interactions in controlled laboratory settings. However, several of these designs are suitable only for studying small model plants such as Arabidopsis thaliana and Brachypodium distachyon and therefore require modification to be extended to larger plant species like crop plants. In addition, specific tools and technical skills needed for fabricating these devices may not be available to researchers. Hence, this study aimed to establish an alternative protocol to generate a larger, modular and reusable plant growth device based on different available resources.
Results
Root-TRAPR (Root-Transparent, Reusable, Affordable three-dimensional Printed Rhizo-hydroponic) system was successfully developed. It consists of two main parts, an internal root growth chamber and an external structural frame. The internal root growth chamber comprises a polydimethylsiloxane (PDMS) gasket, microscope slide and acrylic sheet, while the external frame is printed from a three-dimensional (3D) printer and secured with nylon screws. To test the efficiency and applicability of the system, industrial hemp (Cannabis sativa) was grown with or without exposure to chitosan, a well-known plant elicitor used for stimulating plant defense. Plant root morphology was detected in the system, and plant tissues were easily collected and processed to examine plant biological responses. Upon chitosan treatment, chitinase and peroxidase activities increased in root tissues (1.7- and 2.3-fold, respectively) and exudates (7.2- and 21.6-fold, respectively). In addition, root to shoot ratio of phytohormone contents were increased in response to chitosan. Within 2 weeks of observation, hemp plants exhibited dwarf growth in the Root-TRAPR system, easing plant handling and allowing increased replication under limited growing space.
Conclusion
The Root-TRAPR system facilitates the exploration of root morphology and root exudate of C. sativa under controlled conditions and at a smaller scale. The device is easy to fabricate and applicable for investigating plant responses toward elicitor challenge. In addition, this fabrication protocol is adaptable to study other plants and can be applied to investigate plant physiology in different biological contexts, such as plant responses against biotic and abiotic stresses.
Similar content being viewed by others


Background
In nature, plant roots develop underground and in sophisticated associations with microorganisms, making it challenging to observe root structure and conduct research on root activities [1]. Therefore, several platforms, for example, rhizotrons [2], rhizoponics [3] and rhizoboxes [4], have been developed to facilitate plant root morphological studies in controlled laboratory settings. In addition, technologies like Plant-in-Chip [5], RootChip [6], tracking roots interaction system (TRIS) [7], and ecosystem fabrication (EcoFAB) [8] have been further modified to increase the accessibility of plant-microbe interaction analysis. However, these systems are custom-made, requiring specialized techniques, tools and settings for manufacturing and implementation. Therefore, modification of the designs may be necessary upon the availability of different resources and intended research application.
One of the most recent examples, EcoFAB (https://eco-fab.org/), is an inexpensive and easy-to-fabricate device built based on three-dimensional (3D) printing technology [9, 10]. The original iteration is constructed using a microscope glass slide bonded via a plasma cleaner to custom-built polydimethylsiloxane (PDMS) growth chamber. The PDMS section is cast in a plastic mold, printed from a 3D printer. Optionally, an attachment between the glass slide and the PDMS layer can be reversibly bound using a 3D printed plastic or a machined metal clamp. The EcoFAB model has many benefits. It enables readily accessible observation of root morphology and microbial localization using microscopes and other non-destructive imaging tools. Root biochemical and exudate composition can be collected and analyzed under standardized procedures. The model can use different growth substrates such as soil, sand and liquid [9]. The reproducibility of the EcoFAB device has been verified across multiple laboratories in diverse growth environments [11]. The versatility of the EcoFAB system permits robust studies on model plants such as Arabidopsis thaliana, Brachypodium distachyon and Panicum virgatum. Although appropriate for these model plants, a larger device is required to address research questions in a broader array of plant species like staple and industrial crops, which are generally longer lived and grow to larger sizes than the model plants. Moreover, technical support, including 3D printers, plastic materials and accompanying tools, may vary across different workplaces. Hence, manufacturing processes are dependent upon the availability of the relevant machinery and supplies.
Industrial hemp (Cannabis sativa) is an emerging crop within the agricultural industry worldwide [12]. Its global market is projected to increase from $3.5 billion in 2019 to $18.8 billion in 2025, with a compound annual growth rate of 32.17% [13]. Hemp seed contains low tetrahydrocannabinol (THC) content but a high amount of protein and a good proportion of healthy unsaturated fatty acids [14, 15], creating the demand in the food and beverage industries. Hemp seed oil is a nutritional supplement added to skincare and medicinal products [12, 16]. In addition, hemp fiber is a perfect source for the textile industry owing to its robustness, and high absorbent capacity [17] and hemp hurd has been increasingly processed into hempcrete to replace traditional concrete in construction and building [18].
Despite its benefits, fundamental research to inform and establish daily agronomical practice has been lacking and inconclusive for the growers, who have been unable to transform scientific data into field applications [19]. For example, C. sativa is infected by several pathogenic fungi such as Botrytis cinerea, causing grey mold, Fusarium and Pythium species causing root rot, Macrophomina phaseolina causing charcoal rot, Sclerotinia sclerotiorum causing stem canker and Golovinomyces cichoracearum causing powdery mildew [20,21,22]. These infections suppress plant growth and reduce yield and product quality in outdoor fields and greenhouse settings [21]. However, the pathology underlying the different infections is poorly understood, and disease management programs have not been fully established [22]. The growers may apply inorganic agents, for example, potassium bicarbonate, hydrogen peroxide, boric acid, orthosilicic acid or synthetic fungicides such as fluopyram, to moderate or eradicate fungal pathogens [23]. To avoid using chemicals, natural products such as seaweed extract, plant growth-promoting bacteria, humic substances, and chitin/chitosan derivatives have been used to increase product yield and promote plant defense to combat pests and diseases in other crops [24, 25]. They can be mixed into the soil or diluted and sprayed on aboveground plant tissues [26, 27]. Nonetheless, the benefits of any approach have not yet been comprehensively examined in C. sativa plants. Verifying their stimulating effects will benefit both industrial hemp and medicinal cannabis (high-THC cultivars) industries to tackle fungal disease problems in the field.
As principally inspired by the EcoFAB model, we developed a new device called Root-Transparent, Reusable, Affordable 3D Printed Rhizo-hydroponic or Root-TRAPR system. The device was enlarged and strengthened to cope with industrial hemp growth. To demonstrate the usability and effectiveness of the system, an elicitor challenge assay using colloidal chitosan was developed. Its effect was examined on plant defense by monitoring plant root development and analyzing biological responses by measuring specific enzymatic activities and phytohormone levels. The Root-TRAPR system could be a convenient testing platform for verifying the stimulating effects of plant elicitors on C. sativa plants to further the goals of sustaining and promoting the expanding cannabis industry.
Results
Generation of Root-TRAPR system
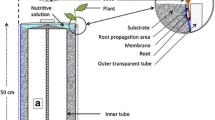
Through a process of iterative design, the Root-TRAPR system was created based on available resources at the University of Melbourne, Australia. The model was inspired by a range of plant growth devices, including the recent EcoFAB model developed at Lawrence Berkeley National Laboratory, US [9]. We retain some elements of the original EcoFAB design, including a glass microscope slide base with a PDMS layer. Differently, our chamber is enclosed by an acrylic sheet, sealed using a compression seal supported by a 3D-printed external structural frame. An exploded-view diagram displaying the components of the Root-TRAPR system and the assembly is shown in Fig. 1. Representatives of the system and all components are featured in Fig. 2, and the details of each part are described in Table 1.

Exploded-view diagram displaying the main components of the Root-TRAPR system. The internal root growth chamber comprises an upper acrylic sheet, a customized PDMS gasket and a bottom microscope slide. The external structural frames (top and bottom) are made of 3D-printed PLA plastic, retained with nylon bolts and nuts (× 8). All components are stable to ethanol, facilitating decontamination and sterilization

Pictures displaying a complete assembled Root-TRAPR system with industrial hemp grown inside (a and b) and all individual parts (c–i) – the internal root growth chamber consisted of an acrylic sheet, a PDMS gasket and a microscope slide (c), top and bottom external structural frames (d), PDMS mold (e), nylon bolts and nuts (f), rubber bung (g), stand (h) and window shutter (i)
The Root-TRAPR system comprises of two major components—an internal root growth chamber (Fig. 2c) and an external structural frame (Fig. 2d). The internal root growth chamber has a transparent viewing configuration from either top or bottom sides through a transparent acrylic sheet and microscope slide, respectively, to facilitate plant root structure observation. A square PDMS gasket with an oval void in the center is pre-cast in a 3D-printed plastic mold (Fig. 2e), enabling fine tuning the void volume by increasing/decreasing the oval width or gasket thickness. The acrylic sheet is pre-drilled with upper and lower holes to insert the plant seed and exchange plant growing media. The elastic PDMS gasket is inserted between the acrylic sheet and microscope slide to create a root growth chamber.
The three internal layers comprised of acrylic sheet, PDMS gasket and microscope slide are secured and compressed using top and bottom external frames printed from a fused deposition modeling (FDM) 3D printer using an inexpensive polylactic acid (PLA) plastic material. The frame is furnished with eight sets of nylon bolts and hexagon nuts (Fig. 2f) to fasten and compress the whole model together tightly. A rubber bung (Fig. 2g) is plugged into the lower smaller hole of the acrylic sheet to stop leakage. During growth experiments, the stand (Fig. 2h) and window shutter (Fig. 2i) can be additionally put in place to tilt the model at a 25° angle to promote gravitropism and prevent direct light onto the plant roots, respectively. The assembled Root-TRAPR device is not damaged by absolute ethanol, therefore the model can be submerged in the solvent for decontamination and sterilization before use.
The approximate cost of the Root-TRAPR system is detailed in Table 1. All 3D-printed objects are subjected to a subsidized AU$0.15 per 1 g material according to the standard printing price for the University of Melbourne [28]. The total cost is approximately AU$64.0 per unit but could vary based on differing plastic materials, printing resolution, machinery techniques or bulk supplies used.
Industrial hemp growth in Root-TRAPR system
Plant growth experiments were carried out using three biological replicates under two different condition—control and chitosan treatment. After germination in Petri dishes, industrial hemp seedlings were transferred to the Root-TRAPR systems and maintained for 14 days in a controlled environment with Hoagland nutrient solution (Additional file 1). After 7 days of growth, nutrient solutions were exchanged. Control plants were treated with standard Hoagland solution and chitosan treatment was performed by substituting plain Hoagland solution with the solution containing 1% w/v colloidal chitosan. Plant growth was monitored with root structure recorded every 2–3 days using a modified scanner connected with the WinRHIZO software. Upon harvest on day 14, plant root and shoot tissues and root exudate were collected and subsequently processed for enzymatic assays, phytohormone quantifications and gene detections.
Root morphology was captured by a well-calibrated optical light scanner and analyzed by the WinRHIZO software throughout the study (Fig. 3). Root growth was monitored through three different parameters—root length, root surface area and average root diameter. Under control conditions, plants constantly expanded their roots throughout 14 days of observation, ending at 55.27 ± 5.06 cm and 12.33 ± 1.35 cm2 in length and surface area, respectively (Fig. 4a–d). The expansion rate was slow during the first week (from 5.47 to 16.49 cm in length and 1.17 to 3.32 cm2 in surface area) but increased during the second week (from 16.49 to 55.27 cm in length and 3.32 to 12.33 cm2 in area). Despite enlarging in root length and surface area, the average root diameter did not change during the monitoring period (0.72 to 0.71 mm; Fig. 4e–f). This indicates that plants expanded existing roots to a larger size and at the same time generated new lateral roots. Young secondary and tertiary branch roots, ranging between 0.2–0.5 mm in diameter, offset the larger primary and pre-existing branch roots (Fig. 3). Therefore, average root diameter of the control plants remained constant.

Photos presented sequential development of industrial hemp in the Root-TRAPR system comparing control and chitosan-treated condition from day 0 to day 14. Chitosan treatment was conducted on day 7 of the experiment. Left-panel pictures of each condition show top-view photos taken on a smartphone camera. Right-panel pictures represent root images captured using a WinRHIZO root scanner

Graphs depicting root developments of industrial hemp in the Root-TRAPR system comparing control and chitosan-treated plants. The measurements are root length (a and b), root surface area (c and d), and average root diameter (e and f). Line graphs (a, c and e) display the root development of three individual replicates of each group. Bar graphs (b, d and f) show an average with an error bar of the standard error of the mean (SEM). Different capital letters above bar graph (A and B) refer to a significant difference at p-value < 0.05 tested by one-way ANOVA, followed by Tukey's HSD test across treatment and day
In the chitosan-treated group, plant roots developed well before chitosan was introduced on day 7, cumulative at 10.98 ± 3.65 cm in total root length and 2.28 ± 0.63 cm2 in root surface area, which were not significantly different from the controls (Fig. 4a–d). However, after the treatment, plants displayed significantly reduced root expansion, finishing at 12.79 cm ± 3.89 in length and 2.91 ± 0.80 cm2 in surface area, which were significantly smaller than those of control plants (p-value = 0.003 and 0.004, respectively). The average root diameter was slightly increased after the treatment, expanding from 0.69 ± 0.05 mm (day 7) to 0.74 ± 0.02 mm (day 14) but was not statistically significant and did not differ from the controls (Fig. 4e–f). As observed from the root morphology (Fig. 3), chitosan-treated plants generated remarkably fewer new branch roots than the controls. This could reflect the slight increase in root diameter of chitosan-treated plants.
The reduction of root expansion after exposure to chitosan is consistent with previous observations on Arabidopsis thaliana [29, 30]. In those studies, plants struggled to elongate roots when exposed to chitosan from as low as 0.01% w/v in concentration. The chitosan disruption on root growth is unlikely to be caused by an increase in viscosity or osmolality of the chitosan suspension. In preparation, the mixture turned into a gel-like suspension after chitosan was added into a Hoagland base liquid. Based on experimental measurements, 1% w/v chitosan is 13.72 times more viscous than water [31] with an osmolality of approximately 94 mOsm/kg in Hoagland solution. The osmolality is calculated from 72 mOsm/kg of chitosan in pure water [31], plus values from other salt ingredients [32]. Typical osmolality in plant cells ranges from 300–700 mOsm/kg [33], hence an increase in osmolality caused by chitosan should have minimal impact on plant nutrient uptake and cellular tonicity. Whereas, increase in viscosity could conceivably contribute to a creation of physical barrier that could limit root growth. However, previous studies have demonstrated that root growth is not impeded by 1% agar solutions. Viscosity of 0.8–1% agar which is normally supplied into plant growth media is approximately 30–80 times higher than water (depending on pH and temperature) [34]. As shown in several morphological studies, plant roots can penetrate through agar media and demonstrate natural growth [30, 35]. Therefore, the reduction of root expansion is likely a direct effect of chitosan on the plant root system.
To assess the health of the plants grown in the Root-TRAPR system, hydrogen peroxide (H2O2) content was measured in root and shoot tissues and compared to the plants grown in a mini hydroponic-like system (Additional file 2). H2O2 is a key signaling molecule in plant response upon environmental stresses. Increase in H2O2 level can be caused by natural factors, such as heat, cold, drought and salinity, resulting in redox imbalance and oxidative stress in plant cells [72]. It could therefore be a potential elicitor to help counteract fungal diseases in agriculture.
Conclusion
In this research, the Root-TRAPR system has been developed to grow larger plants under in vitro conditions. The system was tested using an industrial crop** cultivar of C. sativa, then exploring the impact of chitosan, a potential defense elicitor molecule, on plants. The system enabled the visualization of root material and the ability to harvest plant tissues and exudates that were then successfully processed for enzymatic activity assays, phytohormone measurements and nucleic acid extraction. After treating the plants with 1% w/v chitosan for 7 days, chitinase and peroxidase activities were promoted in root tissues and exudates. This confirms the effect of chitosan to induce plant defense enzymes associated with increased disease resistance. Finally, the Root-TRAPR system has opened the way for further analysis of C. sativa and other larger plants under defined in vitro conditions.
Methods
Chemicals
Analytical grade solvents (ethanol, isopropanol, hydrochloric acid, phosphoric acid) were supplied by ChemSupply, Australia. LC–MS grade solvents (acetonitrile and formic acid) were acquired from Thermo Fisher Scientific, US. Deionized water was used during plant growth experiments, whereas Milli-Q water (Merck Millipore, Germany) was used for all analytical processes. Formulation of Hoagland solution is supplied in Additional file 1.
Root-TRAPR system fabrication
Materials used for fabricating the Root-TRAPR system are listed in Table 1, and the fabrication procedure is described step by step.
Printing PDMS mold, external frames and supplementary parts
PDMS mold (Fig. 2e), top and bottom external structural frame (Fig. 2d), stand (Fig. 2h) and window shutter (Fig. 2i) were manufactured using an FDM 3D printer (MakerBot Replicator plus, US). All components were designed using an open-source computer-aided design (CAD) software (FreeCAD, https://www.freecadweb.org/). The design files are supplied in Additional files 8, 9, 10, 11, 12, 13. The infill was set at 80% for the external structural frame and 10% for PDMS mold, stand and window shutter. The objects were printed on a raft base layer with a light fill support underneath. Layer height was set at 0.2 mm with two shells. After printing, the internal surfaces of the PDMS mold were finished with coarse (P80) and fine (P600) sandpapers. Printing scrap and support were removed from all printed items before use.
Casting PDMS gasket
A Sylgard 184 elastomer kit (Dow Corning, US) created the PDMS gasket. Standard 10:1 (w/w) ratio of base to catalyst was used. Eighteen g of silicone base was mixed with 1.8 g of curing agent in a disposable foil baking cup. The mixture was placed in a vacuum chamber for 30 min to remove air bubbles and then gently poured into the 3D-printed PDMS mold. Overfill was removed by scra** a ruler across the top surface of the mold. The filled mold was incubated at 55 °C overnight to allow the elastomer to set. The solidified PDMS gasket was slowly cut away from the mold using a single edge razor blade. The completed PDMS gasket is shown in the middle of Fig. 2c.
Preparation of the upper viewing window acrylic sheet
An acrylic sheet was cut into the desired size (128 × 85 mm) using a Felder BF-5 combination machine (Felder Group, Austria). Then two circular holes were added using a drill press with appropriately diameter sizes of 8- and 9-mm bits. The upper larger hole (9 mm) was left blank for placing the seed, while the lower smaller hole (8 mm) was firmly stoppered with a rubber bung (Fig. 2g) to stop leakage. A completed acrylic sheet is presented on the left of Fig. 2c.
Assembly of Root-TRAPR unit
The completed Root-TRAPR system was assembled by placing the PMDS gasket between a microscope glass slide underneath and an acrylic sheet on top. The three internal components were then positioned inside the pocket of the bottom external frame and enclosed by the top frame. Finally, eight pre-sized nylon bolts and nuts (Fig. 2f) were screwed in to tighten the layers and complete the main assembly. Additionally, during growth experiments, the stand (Fig. 2h) and window shutter (Fig. 2i) can be incorporated to tilt the model at a 25° angle from the ground to promote gravitropism and prevent direct light onto the plant root, respectively.
Sterilizing Root-TRAPR system
Before use, the assembled Root-TRAPR system and supporting parts were placed in a plastic container and submerged in 70% ethanol for 30 min and 100% ethanol for another 10 min. It was shaken occasionally to ensure all parts were exposed to the solvent, and the oval root growth chamber was filled throughout. After sterilization, the solvent was drained off, and the model was dried in a laminar flow cabinet. Once seedlings had germinated, the sterilized Root-TRAPR system was rinsed with autoclaved deionized water and filled with 15 ml of full-strength Hoagland solution.
Colloidal chitosan preparation
Colloidal chitosan was prepared according to the previous method [73] with a slight modification. Five g of chitosan powder (medium molecular weight; Sigma, US) was first mixed with 50 ml of 85% phosphoric acid, followed by slowly adding another 50 ml of the acid with continuous stirring. The mixture was left at 4 °C overnight to form a colloidal suspension. Pre-cooled 500 ml of 50% ethanol was added to dilute the mixture, then left at 4 °C overnight again. The suspension was filtered through Whatman Grade 1 filter paper (Whatman plc, UK), aided by vacuum filtration. Colloidal chitosan was retained in the funnel and then washed with distilled water until pH above 5. The retentate was transferred to 50-ml conical tubes and then lyophilized in an Alpha 1–4 LD plus freeze-drier (Christ, Germany). Before use, dried chitosan was resuspended to 1% w/v in Hoagland solution.
Seed germination
The overview of experimental workflow starting from seed germination until sample collection is illustrated in Fig. 7. Industrial hemp seeds, Ferimon (France) was received from Southern Hemp Australia. Obtaining and processing industrial hemp (low-THC cannabis) at the University of Melbourne is authorized by Agriculture Victoria, the State Government (authority number 2019/12). The seeds were sterilized with 70% ethanol for 1 min and 0.04% sodium hypochlorite for 10 min, followed by rinsing three times with autoclaved deionized water. Sterile seeds were imbibed at room temperature overnight and transferred to round Petri dishes (90 mm in diameter) containing a moistened filter paper. Germination was conducted in the dark at ambient temperature (approximately 20 °C) for 3 days. Day 0 was counted when the seedlings were transferred into the Root-TRAPR system.
Plant growth and chitosan treatment
Seedlings with 4–6 cm-long tap root were transferred to the Root-TRAPR systems supplied with 15 ml of Hoagland solution using sterilized forceps. Plants were maintained for 7 days in a CMP6010 growth chamber (Conviron, Canada) at 25 °C for 16 h with light and at 21 °C for 8 h of darkness. Light intensity was set at level 2, and relative humidity was maintained at 60%. The nutrient solution was filled up every 2–3 days to compensate for liquid consumption and evaporation. On day 7, plants were separated into control and chitosan conditions. The entire solution was collected and substituted with a new 15 ml of Hoagland solution in the control group. The pre-treated solution was collected and replaced with 1% w/v colloidal chitosan suspension in the chitosan group. All plants were maintained under the same condition for another 7 days. Hoagland solution (approximately 1–2 ml) was added up every 2–3 days in both groups for liquid compensation.
Root growth measurement
Root growth was monitored and analyzed under a well-calibrated root scanning system composed of an optical scanner (Epson Perfection V800, Japan) equipped with WinRHIZO Arabidopsis 2019 software (Regent Instruments, Canada). Plant roots were scanned every 2 to 3 days by placing the Root-TRAPR device straight on the scanner's document table without removing the liquid medium, with the scanner lid open. Root measurement was determined in three different parameters – root length, root surface area and average root diameter. The software automatically detected the root region in a greyscale mode. As a result, the roots are visible as brighter regions than the background. A manual adjustment was carried out when the automatic detection misread any root region. The software automatically measured root parameters using standard precision and normal cross detection settings. Pictures showing how the software detected regions and measured root parameters are displayed in Additional file 14. The outermost area labelled in green refers to the analyzed root region, while small inner areas labelled in red are manually excluded from the analysis. The software used different colors to define different root diameters ranging from 0–0.5 mm until 4.5–5 mm. For example, roots in red color refer to root diameter of 0–0.5 mm, and roots in yellow color are 0.5–1 mm in diameter. The summation of root length from all root sizes is the total root length and the root length multiplied by the root diameter is the whole root surface area. Plant shoot and overview plant structure were also photographed using a smartphone camera (** under the Root-TRAPR root growth chamber. They were ground using mortar and pestle under freezing conditions of liquid nitrogen. Fine tissue powder was separately transferred to three micro-centrifuge tubes in approximate 100 mg by weight (Fig. 7). The tubes were weighed and stored in a -80 °C freezer until further use.
Root exudate was collected twice on day 7 (pre-treatment) and day 14 (post-treatment). First, it was drawn from the Root-TRAPR root growth chamber into a 50-ml conical tube. Next, the solution was spun at 2,500 × g, 4 °C for 20 min to remove debris. Next, the supernatant was transferred to a 10 kDa molecular weight cutoff (MWCO) Amicon Ultra-15 centrifugal filter unit (Merck Millipore, Germany) and then centrifuged at 4,000 × g, 4 °C for 40 min to concentrate root exudate proteins. Approximately 200 µl of protein fraction was captured in the filter unit and stored at -80 °C until further use.
Biological assays
For tissue samples (shoot and root), 1 ml of 100 mM phosphate buffer, pH 6.5, was added to extract proteins from tissue powder (approximately 100 mg). The tube was vortexed and centrifuged at 13,000 × g for 20 min. The supernatant was collected and stored at -20 °C until assay. For root exudate, concentrate protein (approximately 200 µl) was straightaway assayed as follows.
H2O2 detection
The working solution of titanium tetrachloride (TiCl4) was pre-made by slowly adding 100 µl of concentrated TiCl4 solution (product code: 208566, Sigma, US) to 100 µl of 6 M hydrochloric acid (HCl) on ice. The mixture was left at 4 °C overnight and then diluted in 10 ml of 6 M HCl.
Twenty µl of tissue extract was mixed with 80 µl of 100 mM phosphate buffer, pH 6.5 in a 96-well microplate. Immediately before detection, 100 µl of working TiCl4 solution was added to each well. Absorbance was measured at 415 nm using an EnSpire Multilabel plate reader (PerkinElmer, US). H2O2 content was calculated against a calibration curve created from serial dilutions of 0.001–0.05% v/v standards.
Protein measurement
Bradford reagent (Bio-Rad, US) was diluted five times in deionized water. A 20 µl of protein extract was mixed with 180 µl of diluted Bradford reagent in a 96-well microplate. The mixture was incubated at room temperature for 10 min. Absorbance was detected at 595 nm using the plate reader. Protein concentration was measured against a bovine serum albumin (BSA) standard curve (0–100 µg/ml).
Peroxidase activity
Twenty µl of protein extract was mixed with 150 µl of 0.025% H2O2, diluted in 100 mM phosphate buffer, pH 6.5 in a 96-well microplate. Immediately before the assay, 50 µl of 50 mM guaiacol was added to the solution. Absorbance was measured at 470 nm and repeated every 30 s. The rate of absorbance change on the first 3 min was calculated to represent guaiacol peroxidase activity in a unit of ΔOD/min, normalized to protein amount.
Chitinase activity
Dimethylaminobenzaldehyde (DMAB) stock solution was prepared by dissolving 8 g of DMAB pellet in a mixture of 70 ml of glacial acetic acid and 10 ml of 32% HCl. Before the assay, a working DMAB solution was prepared by diluting the stock solution ten times in glacial acetic acid.
Forty µl of protein solution was mixed with 100 µl of 1% w/v of colloidal chitin [74], suspended in 50 mM acetate buffer, pH 5.5 and then incubated at 37 °C for 2 h. The reaction was stopped by centrifugation at 8,000 × g for 10 min. Forty µl of 1 M sodium borate buffer, pH 8.5, was added into a mixture, then incubated at 95 °C for 5 min and cooled on ice for 20 min. Five hundred µl of working DMAB reagent was added into a solution, then incubated at 37 °C for 20 min. The final solution was aliquoted into a 96-well microplate and detected at 585 nm. Chitinase activity was evaluated against GlcNAc standard curve (0.02–2 mM) and expressed as mmole GlcNAc released per 1 g protein.
Phytohormone measurement
Phytohormones were extracted from tissue powder using 200 µl of 70% methanol supplied with 500 ng/ml of internal standards (d5-zeatin, d2-IAA, d7-CA, d4-SA, d6-ABA and H2JA). Samples were vortexed and centrifuged at 13,000 × g for 20 min. The supernatant was transferred into a glass LC–MS vial containing an insert and injected into a 1200 series LC system equipped with a 6410 Triple Quadrupole MS machine (Agilent, US). Metabolites were separated on Eclipse XDB-C18, 1.8 µm, 2.1 × 100 mm column (Agilent, US). The column temperature was set at 45 °C. Mobile phase A and B were 0.1% formic acid in water and acetonitrile, respectively. The elution gradient was set as follows: 80% A (0–2 min), 80–50% A (2–3 min), 50–5% A (3–12 min), 5% A (12–16 min), 5–80% A (16–17 min) and 80% A (17–23 min). The flow rate was 320 µl/min, and the injection volume was 5 µl. Analytes were ionized using electrospray ionization (ESI) source with capillary voltage at 5500 V and 4500 V for positive and negative modes, respectively. The nebulizer was set at 55 psi. Nitrogen gas flow was maintained at 13 L/min and 250 °C. According to the published method [75], phytohormones were detected using multiple reaction monitoring (MRM) program. The MRM transitions, collision energies and polarities were applied as follows: zeatin (220.1 → 136.1 m/z, 14 eV, positive), IAA (176.1 → 130.1 m/z, 10 eV, positive), CA (149.1 → 103.1 m/z, 20 eV, positive), BL (481.5 → 315.3 m/z, 10 eV, positive), SA (137.0 → 93.0 m/z, 16 eV, negative), ABA (263.1 → 153.1 m/z, 8 eV, negative), JA (209.1 → 59.0 m/z, 8 eV, negative), JA-Ile (322.1 → 129.9 m/z, 24 eV, negative), OPDA (291.0 → 164.9 m/z, 20 eV, negative), d5-zeatin (225.2 → 137.1 m/z, 20 eV, positive), d2-IAA (178.1 → 132.0 m/z, 12 eV, positive), d7-CA (156.1 → 109.0 m/z, 22 eV, positive), d4-SA (141.0 → 97.1 m/z, 16 eV, negative), d6-ABA (269.1 → 159.1 m/z, 8 eV, negative) and H2JA (211.1 → 59.0 m/z, 12 eV, negative). Phytohormone concentrations were measured by comparing relative peak area against calibration curves created from serial dilutions of the standards. The curve was plotted from 4–6 data points in a range of 10–1000 ng/ml according to the phytohormone levels found in the samples.
DNA extraction and PCR analysis
Four hundred µl of DNA extraction buffer (160 mM Tris, 56 mM EDTA, 30 mM sodium metabisulfite and 1.6 M sodium chloride) was added into tissue powder (approximately 100 mg) and centrifuged at 13,000 × g for 5 min. Three hundred µl of supernatant was taken and mixed with 300 µl of 100% isopropanol. The mixture was incubated at room temperature for 10 min with occasionally tube-inverting and then centrifuged at 13,000 × g for 5 min. The pellet was washed with 300 µl of 70% ethanol and air-dried overnight. The dried DNA pellet was dissolved in 50 µl of nuclease-free water (Qiagen, Germany). DNA concentration was measured using a UV5Nano spectrophotometer (Mettler-Toledo, US).
Six C. sativa genes (encoding actin, ubiquitin, EF-1α, chitinase 5, chitinase 2 and chitinase 4-like) were predicted from the C. sativa draft genome [55]. Gene and primer details are described in Additional file 7. A 100 ng of DNA template was added to 25 µl of PCR reaction mixture, consisted of 1 × MyTaq Red buffer, 0.5 U MyTaq DNA polymerase (Bioline, US) and 0.4 µM forward and reverse primers each. The PCR amplification was performed using a T100 thermal cycler (Bio-Rad, US) with an initial denaturation of 2 min at 95 °C, followed by 35 cycles of 30 s at 95 °C, 30 s at 55 °C and 1.15 min at 72 °C, and a final extension of 5 min at 72 °C. A 10 µl of the amplification product was resolved in 1% agarose gel electrophoresis at 85 V for 50 min. The gel was stained with ethidium bromide and analyzed using Gel Doc EZ imager equipped with ImageLab software (Bio-Rad, US).
Statistical analysis
A two-tailed student's T-test was used for enzymatic activity and phytohormone content with Microsoft Excel 2016 software. One-way ANOVA followed by Tukey's honestly significant difference (HSD) analysis was used for root growth measurement and hydrogen peroxide content with Minitab 19 software. A p-value below 0.05 was considered as a significant difference between tested conditions. Online MetaboAnalyst 5.0 software [76] was used to perform principal component analysis (PCA) of overall phytohormone content. Before the analysis, Pareto data scaling was employed to normalize shoot tissue data while the data of root tissue was log-transformed and scaled using mean centering.
(STL files can be opened using FreeCAD software, https://www.freecadweb.org/).
Availability of data and materials
All data generated from this study are included in this published article and supporting materials. Additional details on the Root-TRAPR system fabricating procedures can be acquired from the corresponding author upon reasonable request.
Abbreviations
- 3D:
-
Three-dimensional
- ABA:
-
Abscisic acid
- ANOVA:
-
Analysis of variance
- BL:
-
Brassinolide
- CA:
-
Cinnamic acid
- DMAB:
-
Dimethylaminobenzaldehyde
- EcoFAB:
-
Ecosystem fabrication
- EDTA:
-
Ethylenediamine-tetra-acetic acid
- EF-1α:
-
Elongation factor-1 alpha
- FDM:
-
Fused deposition modelling
- FW:
-
Fresh weight
- GlcNAc:
-
N-Acetylglucosamine
- H2O2 :
-
Hydrogen peroxide
- HCl:
-
Hydrochloric acid
- HSD:
-
Honestly significant difference
- IAA:
-
Indole-3-acetic acid
- JA:
-
Jasmonic acid
- JA-Ile:
-
Jasmonyl-isoleucine
- LC:
-
Liquid chromatography
- MRM:
-
Multiple reaction monitoring
- MS:
-
Mass spectrometry
- OD:
-
Optical density
- OPDA:
-
12-Oxo-phytodienoic acid
- PCA:
-
Principal component analysis
- PCR:
-
Polymerase chain reaction
- PDMS:
-
Polydimethylsiloxane
- PLA:
-
Polylactic acid
- Root-TRAPR:
-
Root-transparent reusable affordable three-dimensional printed rhizo-hydroponic
- SA:
-
Salicylic acid
- SEM:
-
Standard error of the mean
- THC:
-
Tetrahydrocannabinol
- TiCl4 :
-
Titanium tetrachloride
References
Ryan PR, Delhaize E, Watt M, Richardson AE. Plant roots: understanding structure and function in an ocean of complexity. Ann Bot. 2016;118(4):555–9.
Taylor HM, Upchurch DR, McMichael BL. Applications and limitations of rhizotrons and minirhizotrons for root studies. Plant Soil. 1990;129:29–35.
Mathieu L, Lobet G, Tocquin P, Perilleux C. “Rhizoponics”: a novel hydroponic rhizotron for root system analyses on mature Arabidopsis thaliana plants. Plant Methods. 2015;11:3.
Oburger E, Dell‘mour M, Hann S, Wieshammer G, Puschenreiter M, Wenzel WW. Evaluation of a novel tool for sampling root exudates from soil-grown plants compared to conventional techniques. Environ Exp Bot. 2013;87:235.
Parashar A, Pandey S. Plant-in-Chip: Microfluidic system for studying root growth and pathogenic interactions in Arabidopsis. Appl Phys Lett. 2011;98:263703.
Grossmann G, Guo WJ, Ehrhardt DW, Frommer WB, Sit RV, Quake SR, et al. The RootChip: an integrated microfluidic chip for plant science. Plant Cell. 2011;23(12):4234–40.
Massalha H, Korenblum E, Malitsky S, Shapiro OH, Aharoni A. Live imaging of root-bacteria interactions in a microfluidics setup. Proc Natl Acad Sci U S A. 2017;114(17):4549–54.
Zengler K, Hofmockel K, Baliga NS, Behie SW, Bernstein HC, Brown JB, et al. EcoFABs: advancing microbiome science through standardized fabricated ecosystems. Nat Methods. 2019;16(7):567–71.
Gao J, Sasse J, Lewald KM, Zhalnina K, Cornmesser LT, Duncombe TA, et al. Ecosystem fabrication (EcoFAB) protocols for the construction of laboratory ecosystems designed to study plant-microbe interactions. J Vis Exp. 2018. https://doi.org/10.3791/57170(134).
Gao J, Northen TR, Lewald K, Cornmesser LT, Andeer PF, inventors; The Regents of the University of California, assignee. Ecosystem for determining plant-microbe interactions. US patent 10787639. 2020 September 29.
Sasse J, Kant J, Cole BJ, Klein AP, Arsova B, Schlaepfer P, et al. Multilab EcoFAB study shows highly reproducible physiology and depletion of soil metabolites by a model grass. New Phytol. 2019;222(2):1149–60.
Schluttenhofer C, Yuan L. Challenges towards revitalizing hemp: a multifaceted crop. Trends Plant Sci. 2017;22(11):917–29.
Valuates Reports. The global industrial hemp market to grow USD 18,812.81 million by 2025, at a CAGR of 32.17%. In: Market research reports. 360iResearch. 2020. https://reports.valuates.com/market-reports/360I-Auto-1N254/the-global-industrial-hemp. Accessed 23 April 2021.
Callaway JC. Hempseed as a nutritional resource: an overview. Euphytica. 2004;140:65–72.
Kleinhenz MD, Magnin G, Ensley SM, Griffin JJ, Goeser J, Lynch E, et al. Nutrient concentrations, digestibility, and cannabinoid concentrations of industrial hemp plant components. Appl Animal Sci. 2020;36(4):489–94.
Gotter A. Hemp oil for skin. Healthline website. 2019. https://www.healthline.com/health/hemp-oil-for-skin. Accessed 23 April 2021.
Duque Schumacher AG, Pequito S, Pazour J. Industrial hemp fiber: a sustainable and economical alternative to cotton. J Clean Prod. 2020;268:122180.
Dhakal U, Berardi U, Gorgolewski M, Richman R. Hygrothermal performance of hempcrete for Ontario (Canada) buildings. J Clean Prod. 2017;142:3655–64.
AgriFutures Australia. Industrial hemp. AgriFutures Australia website. 2017. https://www.agrifutures.com.au/farm-diversity/industrial-hemp/. Accessed 23 April 2021.
McPartland JM, Clarke RC, Watson DP. Hemp diseases and pests: management and biological control. Oxford: CABI Publishing; 2000.
Punja ZK, Collyer D, Scott C, Lung S, Holmes J, Sutton D. Pathogens and molds affecting production and quality of Cannabis sativa L. Front Plant Sci. 2019;10:1120.
Punja ZK. Emerging diseases of Cannabis sativa and sustainable management. Pest Manag Sci. 2021. https://doi.org/10.1002/ps.6307.
Scott C, Punja ZK. Evaluation of disease management approaches for powdery mildew on Cannabis sativa L. (marijuana) plants. Can J Plant Pathol. 2020. https://doi.org/10.1080/07060661.2020.1836026.
Yakhin OI, Lubyanov AA, Yakhin IA, Brown PH. Biostimulants in plant science: a global perspective. Front Plant Sci. 2016;7:2049.
du Jardin P. Plant biostimulants: definition, concept, main categories and regulation. Sci Hortic. 2015;196:3–14.
Debode J, De Tender C, Soltaninejad S, Van Malderghem C, Haegeman A, Van der Linden I, et al. Chitin mixed in potting soil alters lettuce growth, the survival of zoonotic bacteria on the leaves and associated rhizosphere microbiology. Front Microbiol. 2016;7:565.
Olivares FL, Aguiar NO, Rosa RCC, Canellas LP. Substrate biofortification in combination with foliar sprays of plant growth promoting bacteria and humic substances boosts production of organic tomatoes. Sci Hortic. 2015;183:100–8.
NExT Lab. 3D printing at the NExT lab. Melbourne School of Design (University of Melbourne) website. 2021. https://msd-makerspaces.gitbook.io/next-lab/3d-printing/3dp-introduction. Accessed 23 April 2021.
Iglesias MJ, Colman SL, Terrile MC, Paris R, Martin-Saldana S, Chevalier AA, et al. Enhanced properties of chitosan microparticles over bulk chitosan on the modulation of the auxin signaling pathway with beneficial impacts on root architecture in plants. J Agric Food Chem. 2019;67(25):6911–20.
Lopez-Moya F, Escudero N, Zavala-Gonzalez EA, Esteve-Bruna D, Blazquez MA, Alabadi D, et al. Induction of auxin biosynthesis and WOX5 repression mediate changes in root development in Arabidopsis exposed to chitosan. Sci Rep. 2017;7(1):16813.
Batubara I, Rahayu D, Mohamad K, Prasetyaningtyas WE. Leydig cells encapsulation with alginate-chitosan: optimization of microcapsule formation. JEAS. 2012;02(02):15–20.
Rasouli M. Basic concepts and practical equations on osmolality: biochemical approach. Clin Biochem. 2016;49(12):936–41.
Lintunen A, Paljakka T, Jyske T, Peltoniemi M, Sterck F, von Arx G, et al. Osmolality and non-structural carbohydrate composition in the secondary phloem of trees across a latitudinal gradient in Europe. Front Plant Sci. 2016;7:726.
Dyshlyuk L, Dyshlyuk L, Asyakina L, Asyakina L. Study of viscosity of aqueous solutions of natural polysaccharides. Sci Evol. 2016;1(2):11–9.
Aziz AA, Lim KB, Rahman EKA, Nurmawati MH, Zuruzi AS. Agar with embedded channels to study root growth. Sci Rep. 2020;10(1):14231.
Hossain MA, Bhattacharjee S, Armin SM, Qian P, **n W, Li HY, et al. Hydrogen peroxide priming modulates abiotic oxidative stress tolerance: insights from ROS detoxification and scavenging. Front Plant Sci. 2015;6:420.
Asaeda T, Jayasanka SMDH, **a L-P, Barnuevo A. Application of hydrogen peroxide as an environmental stress indicator for vegetation management. Engineering. 2018;4(5):610–6.
Velikova V, Loreto F. On the relationship between isoprene emission and thermotolerance in Phragmites australis leaves exposed to high temperatures and during the recovery from a heat stress. Plant Cell Environ. 2005;28:318–27.
Chaparzadeh N, D’Amico ML, Khavari-Nejad RA, Izzo R, Navari-Izzo F. Antioxidative responses of Calendula officinalis under salinity conditions. Plant Physiol Biochem. 2004;42(9):695–701.
Cheeseman JM. Hydrogen peroxide concentrations in leaves under natural conditions. J Exp Bot. 2006;57(10):2435–44.
Gratani L. Plant phenotypic plasticity in response to environmental factors. Adv Bot. 2014;2014:1–17.
Zakaria NI, Ismail MR, Awang Y, Megat Wahab PE, Berahim Z. Effect of root restriction on the growth, photosynthesis rate, and source and sink relationship of chilli (Capsicum annuum L.) grown in soilless culture. Biomed Res Int. 2020;2020:2706937.
Ferreira RB, Monteiro S, Freitas R, Santos CN, Chen Z, Batista LM, et al. The role of plant defence proteins in fungal pathogenesis. Mol Plant Pathol. 2007;8(5):677–700.
Pusztahelyi T. Chitin and chitin-related compounds in plant-fungal interactions. Mycology. 2018;9(3):189–201.
Bari R, Jones JD. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69(4):473–88.
Shigenaga AM, Argueso CT. No hormone to rule them all: interactions of plant hormones during the responses of plants to pathogens. Semin Cell Dev Biol. 2016;56:174–89.
Steenackers W, El Houari I, Baekelandt A, Witvrouw K, Dhondt S, Leroux O, et al. cis-Cinnamic acid is a natural plant growth-promoting compound. J Exp Bot. 2019;70(21):6293–304.
McAdam SA, Brodribb TJ, Ross JJ. Shoot-derived abscisic acid promotes root growth. Plant Cell Environ. 2016;39(3):652–9.
De Smet I, Signora L, Beeckman T, Inze D, Foyer CH, Zhang H. An abscisic acid-sensitive checkpoint in lateral root development of Arabidopsis. Plant J. 2003;33:543–55.
De Smet I, Zhang H, Inze D, Beeckman T. A novel role for abscisic acid emerges from underground. Trends Plant Sci. 2006;11(9):434–9.
Li X, Chen L, Forde BG, Davies WJ. The biphasic root growth response to abscisic acid in Arabidopsis involves interaction with ethylene and auxin signalling pathways. Front Plant Sci. 2017;8:1493.
Harris JM. Abscisic acid: hidden architect of root system structure. Plants (Basel). 2015;4(3):548–72.
Obianom C, Romanazzi G, Sivakumar D. Effects of chitosan treatment on avocado postharvest diseases and expression of phenylalanine ammonia-lyase, chitinase and lipoxygenase genes. Postharvest Biol Technol. 2019;147:214–21.
Coqueiro DS, de Souza AA, Takita MA, Rodrigues CM, Kishi LT, Machado MA. Transcriptional profile of sweet orange in response to chitosan and salicylic acid. BMC Genomics. 2015;16:288.
van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, et al. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011;12:R102.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134.
Hadwiger LA, Tanaka K. DNA damage and chromatin conformation changes confer nonhost resistance: a hypothesis based on effects of anti-cancer agents on plant defense responses. Front Plant Sci. 2018;9:1056.
Sathya U, Nithya M, Keerthi. Fabrication and characterisation of fine-tuned Polyetherimide (PEI)/WO3 composite ultrafiltration membranes for antifouling studies. Chem Phys Lett. 2020;744:137201.
Kumar A, Yap WT, Foo SL, Lee TK. Effects of sterilization cycles on PEEK for medical device application. Bioengineering. 2018;5:18.
Tee YL, Peng C, Pille P, Leary M, Tran P. PolyJet 3D printing of composite materials: experimental and modelling approach. JOM. 2020;72(3):1105–17.
Pichyangkura R, Chadchawan S. Biostimulant activity of chitosan in horticulture. Sci Hortic. 2015;196:49–65.
Kim H-J, Chen F, Wang X, Rajapakse NC. Effect of chitosan on the biological properties of sweet basil (Ocimum basilicum L.). J Agric Food Chem. 2005;53:3696–701.
de Jail NG, Luiz C, da Rocha Neto AC, Di Piero RM. High-density chitosan reduces the severity of bacterial spot and activates the defense mechanisms of tomato plants. Trop Plant Pathol. 2014;39(6):434–41.
Chien P-J, Sheu F, Yang F-H. Effects of edible chitosan coating on quality and shelf life of sliced mango fruit. J Food Eng. 2007;78(1):225–9.
Hadwiger LA. Multiple effects of chitosan on plant systems: solid science or hype. Plant Sci. 2013;208:42–9.
Gong BQ, Wang FZ, Li JF. Hide-and-seek: chitin-triggered plant immunity and fungal counterstrategies. Trends Plant Sci. 2020;25(8):805–16.
Yin H, Du Y, Dong Z. Chitin oligosaccharide and chitosan oligosaccharide: two similar but different plant elicitors. Front Plant Sci. 2016;7:522.
Lopez-Moya F, Suarez-Fernandez M, Lopez-Llorca LV. Molecular mechanisms of chitosan interactions with fungi and plants. Int J Mol Sci. 2019;20(2):332.
Malerba M, Cerana R. Chitosan effects on plant systems. Int J Mol Sci. 2016;17(7):996.
Suarez-Fernandez M, Marhuenda-Egea FC, Lopez-Moya F, Arnao MB, Cabrera-Escribano F, Nueda MJ, et al. Chitosan induces plant hormones and defenses in tomato root exudates. Front Plant Sci. 2020;11:572087.
Berens ML, Berry HM, Mine A, Argueso CT, Tsuda K. Evolution of hormone signaling networks in plant defense. Annu Rev Phytopathol. 2017;55:401–25.
Sharp R. A review of the applications of chitin and its derivatives in agriculture to modify plant-microbial interactions and improve crop yields. Agronomy. 2013;3(4):757–93.
Olicon-Hernandez DR, Vazquez-Landaverde PA, Cruz-Camarillo R, Rojas-Avelizapa LI. Comparison of chito-oligosaccharide production from three different colloidal chitosans using the endochitonsanolytic system of Bacillus thuringiensis. Prep Biochem Biotechnol. 2017;47(2):116–22.
Shen CR, Chen YS, Yang CJ, Chen JK, Liu CL. Colloid chitin azure is a dispersible, low-cost substrate for chitinase measurements in a sensitive, fast, reproducible assay. J Biomol Screen. 2010;15(2):213–7.
Cao D, Lutz A, Hill CB, Callahan DL, Roessner U. A quantitative profiling method of phytohormones and other metabolites applied to barley roots subjected to salinity stress. Front Plant Sci. 2016;7:2070.
Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46(W1):W486–94.
Acknowledgements
We thank David Brian from Southern Hemp and Ken Stuckey from Leawood Hemp for supplying industrial hemp seeds, Michael Zammit from Science Workshop, the University of Melbourne for preparing acrylic sheets, and Gary Mather from Engineering Workshop and Eric Jong from Research Computing Service Training at the University of Melbourne for providing training and suggestion on 3D printing techniques. We also thank teams in the 3D Innovation Centre, Faculty of Engineering and Information Technology, and the New Experimental Technology Lab (NExT Lab), Melbourne School of Design, for assisting with 3D printing. In addition, Joshua Heazlewood and Ute Roessner provided suggestions on experimental workflow.
Funding
This study was funded by the School of BioSciences Seed Funding for 2019 (SEED19), University of Melbourne. AI had support from the Australian Research Council (LP170100548). PS received a Melbourne Research Scholarship (University of Melbourne) and additional project funding through Nutrifield Pty Ltd.
Author information
Authors and Affiliations
Contributions
PS, RW, and BAB designed and planned the study. PS conducted the experiments and analyzed the data. RW, AI, JSP and BAB provided guidance and technical support throughout the study. PS prepared the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and agree to publish the paper.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
The formula of the Hoagland solution used for growing industrial hemp in this study.
Additional file 2:
Comparison of industrial hemp after 2 weeks growth in the Root-TRAPR systems compared to a mini hydroponic-like system (50-ml conical tube).
Additional file 3:
Two-week-old industrial hemp plants grown in potting soil. They grew taller and bigger than the plants grown in the Root-TRAPR systems, but both developed the same number of leaves and nodes.
Additional file 4:
Summarized protein content, peroxidase and chitinase activities from shoot and root tissues and pre- and post-exudate compared between control and chitosan conditions.
Additional file 5:
Boxplots showing phytohormone levels in separate graphs compared between control and chitosan conditions.
Additional file 6:
Principal component analysis (PCA) of phytohormone contents comparing control and chitosan-treated plants in shoot and root tissues.
Additional file 7:
Gene and primer details of six C. sativa genes detected in this study.
Additional file 8:
3D design file of PDMS mold.
Additional file 9:
3D design file of the bottom external structural frame.
Additional file 10:
3D design file of the top external structural frame.
Additional file 11:
3D design file of the stand.
Additional file 12:
3D design file of left window shutter.
Additional file 13:
3D design file of right window shutter.
Additional file 14:
Pictures showing how WinRHIZO software detects root areas and analyzes root parameters.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Suwanchaikasem, P., Idnurm, A., Selby-Pham, J. et al. Root-TRAPR: a modular plant growth device to visualize root development and monitor growth parameters, as applied to an elicitor response of Cannabis sativa. Plant Methods 18, 46 (2022). https://doi.org/10.1186/s13007-022-00875-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-022-00875-1