
Abstract
Background
Rapid habitat loss and degradation are responsible for population decline in a growing number of species. Understanding the natural history of these species is important for designing conservation strategies, such as habitat enhancements or ex-situ conservation. The acquisition of observational data may be difficult for rare and declining species, but metagenomics and metabarcoding can provide novel kinds of information. Here we use these methods for analysing fecal samples from an endangered population of a colobine primate, the banded leaf monkey (Presbytis femoralis).
Results
We conducted metagenomics via shotgun sequencing on six fecal samples obtained from a remnant population of P. femoralis in a species-rich rainforest patch in Singapore. Shotgun sequencing and identification against a plant barcode reference database reveals a broad dietary profile consisting of at least 53 plant species from 33 families. The diet includes exotic plant species and is broadly consistent with > 2 years of observational data. Metagenomics identified 15 of the 24 plant genera for which there is observational data, but also revealed at least 36 additional species. DNA traces for the diet species were recovered and identifiable in the feces despite long digestion times and a large number of potential food plants within the rainforest habitat (>700 species). We also demonstrate that metagenomics provides greater taxonomic resolution of food plant species by utilizing multiple genetic markers as compared to single-marker metabarcoding. In addition, full mitochondrial genomes of P. femoralis individuals were reconstructed from fecal metagenomic shotgun reads, showing very low levels of genetic diversity in the focal population, and the presence of gut parasites could also be confirmed. Metagenomics thus allows for the simultaneous assessment of diet, population genetics and gut parasites based on fecal samples.
Conclusions
Our study demonstrates that metagenomic shotgun sequencing of fecal samples can be successfully used to rapidly obtain natural history data for understudied species with a complex diet. We predict that metagenomics will become a routinely used tool in conservation biology once the cost per sample reduces to ~100 USD within the next few years.
Similar content being viewed by others

Background
Human impacts on the environment are responsible for a dramatic increase in habitat destruction and an ever increasing list of species that are in decline. For example, most species of mammals have lost more than half of their original range since the 19th century [1] and for well-studied mammals such as primates over half of the species are listed as endangered [2]. Moreover, rapid habitat loss is responsible for extinction rates that have been estimated to be over 100 times higher than the background rates [3]. The decline of some species can be slowed through conservation measures such as habitat preservation, enhancement or ex-situ management, but such measures require natural history data on the fundamental aspects of the species’ biology, distribution, and genetic diversity. The need for such information is urgent [4], but coincides with the decline of natural history research [5, 6]. Furthermore, the limited resources available are disproportionally spent on a few charismatic species thus leaving little funding for other species [7, 8]. Yet these are likely to represent the majority of the endangered species and populations [9]. There is thus a need for develo** new techniques capable of rapidly expanding the data that are obtained in limited field studies often applied to such species.
Valuable natural history information can be obtained by the in-depth study of non-invasive samples such as feces, even if available in small numbers only. Fecal samples are often collected opportunistically during routine field work or they can be obtained efficiently using detection dogs [10]. Such samples have the potential to simultaneously provide information on host genetics, diet, and intestinal parasites. Some of this information can be obtained by direct morphological examination of fecal samples, e.g. by studying diet remnants [11] and gut parasites [12]. Molecular methods have expanded the utility of fecal samples by allowing the analysis of host genetics [13], diet from various sources [14] and the detection of parasites [15]. However molecular methods have been labour intensive as the characterization of multiple species from complex samples involved cloning and Sanger sequencing [14]. The advent of high-throughput sequencing (HTS) has simplified the characterization of complex fecal DNA and now allows for simultaneous characterization of the different aspects of ecology of a species [16]. For fecal samples, HTS can be employed in two ways, either by direct shotgun sequencing of DNA extracted from the fecal samples (metagenomics) or by PCR-based metabarcoding of target genes.
Currently, metabarcoding is more widely used [17, 18], and it has an advantage of lower cost where large numbers of samples have to be screened. Metagenomic shotgun sequencing, in contrast, remains largely unexplored for use in conservation biology [16, 19]. This is presumably due to the higher cost of sequencing and the greater bioinformatics effort required for analysing metagenomic data. But this approach has the potential advantage of rapidly yielding data on genetics, diet, parasites and microbiota from fecal samples, while also avoiding the need for a priori selection of amplification targets which limits the study to the sequencing of a specific subset of the genetic material [20]. This makes metagenomics attractive, but also raises practical challenges. Firstly, bioinformatic challenges arise from the need for a comprehensive reference database against which shotgun data can be queried [16]. Secondly, because of its costs shotgun sequencing is mostly suitable for studies requiring few samples, although with the expectation of cheaper DNA sequencing, one could argue that now is a good time to evaluate and develop the bioinformatic tools for metagenomic data.
The critically endangered population of banded leaf monkeys (Presbytis femoralis femoralis) in Singapore is one case where field observational data has been particularly difficult to obtain. Initially described from Singapore and common in the 19th century, the only remaining population now comprises ~40 individuals that are restricted to the Central Catchment Nature Reserve [21]. The forest is surrounded by urban areas and affected by further urban development, which creates conservation challenges including habitat loss, fragmentation and direct anthropogenic disturbance. The situation is exacerbated by the low genetic diversity within the population [22]. Studies of the species’ autecology, prior to develo** conservation strategies, have been hampered by the difficulty of making direct observations; a 6-month study in the 1990s led to only 13 sightings [23]. Overall, our current understanding of the species biology is preliminary and here fecal samples can be useful in complementing the current research.
In this study we aim to characterize fecal samples of P. femoralis using metagenomics and metabarcoding, for comparisons with field observational data on feeding ecology. Our recent pilot study comparing these approaches for diet analyses in the red-shanked doucs (Pygathrix nemaeus) [16] in a controlled zoo environment suggested that shotgun sequencing yields better taxonomic resolution if utilizing multiple reference loci, as compared to single marker metabarcoding, but this was at the expense of lower detection probability of rare food plants in the sample. Here we increased the depth of shotgun sequencing to obtain high taxonomic resolution whilst also detecting rare diet items. We also test whether DNA based analyses are congruent with field observational data, given that this is the first study applying metagenomics to samples collected in the wild. The challenges are considerable because colobine primates have long digestion times that may cause high DNA degradation (Mean Retention Time >40 h [24]), plant barcodes are short and often not species-specific [25], the potential diet of banded leaf monkeys consists of >700 species of trees and lianas in the studied habitat [26], and the amount of target DNA is minute in comparison to the DNA of microbial origin in fecal material. Despite these difficulties, we show that fecal samples can yield a credible set of well-identified plant sequences that correlates with field observational data. In addition, shotgun sequencing provides data on population genetic structure and gut parasites of individual monkeys.
Results
Field observations
Two and a half years of field observations yielded 31 feeding observations and banded leaf monkeys were seen to feed on 27 plant species from 24 genera and 20 families during the surveys (Additional file 1: Table S2, Table 1). Diet was primarily comprised of fruits and leaves, and to a lesser degree of flowers. Of the 27 species, Fibraurea tinctoria, Xanthophyllum ellipticum, Prunus polystachya and Hevea brasiliensis had two feeding observations each, while feeding on all other species was observed only on a single occasion (Additional file 1: Table S2).
Illumina sequencing
Illumina sequencing using HiSeq produced ~67 to ~108 million reads while MiSeq produced ~23 to ~29 million reads per end per sample. For metabarcoding 272,103 to 419,407 sequences per sample were generated for the widely-used marker, P6 loop of trnL. These sequences were subsequently filtered and subjected to variant calling and diet identification.
Diet analysis
BLAST searches of HiSeq and MiSeq metagenomic data were conducted against the plant barcode databases comprising of rbcL, matK and trnL-F sequences from GenBank and newly sequenced data from the Nee Soon Swamp forest. These yielded between 2616 and 6416 sequence reads (0.004–0.008 %) per sample that could be used for taxonomic classification (Additional file 1: Table S4). A large proportion of the shotgun reads could be classified at least to family (87.0–96.2 %) and a substantial proportion had a genus name associated with them (45.0–56.5 %). A smaller fraction of the reads could be identified to species (27.0–39.5 %), i.e., the reads had similarly high matches to multiple species in a genus or family and thus could be identified only to higher taxonomic ranks (Additional file 1: Table S5). For metabarcoding, after applying the different filtering criteria (FC1 [16], variant calling) we retained the following number of unique sequences per sample: 31 (BLM1), 40 (BLM2), 31 (BLM3), 19 (BLM4), 61 (BLM5) and 46 (BLM6). Here, 4.9–15.8 % of the unique sequences produced species level identifications and 13.1–27.5 % were informative to genus-level, while most contained only family-level information (60.7–73.7 %) (Additional file 1: Table S5).
Comparison of metagenomics and metabarcoding
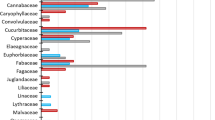
Family-level identifications were largely congruent between the metagenomic (MG) and metabarcoding (MB) analyses (Fig. 1). Metagenomics yielded identifications for 11–25 families per fecal sample (total number of family-level identifications = 99), while metabarcoding revealed 11–22 families per sample (total number of family-level identifications = 93). The use of 95 % or 90 % identity thresholds led to negligible differences for the metabarcoding results (91 vs. 93 identifications). The performance of the two approaches differed at the genus- and species levels (Fig. 1), as metagenomics generated ~2–3 times more identifications at both taxonomic hierarchies (genus: MG total = 115, range = 11–36 vs MB total = 46, range = 4–11; species: MG total = 59, range 3–21 vs MB total = 24, range = 2–7).

Identifications at different taxonomic hierarchies using metagenomics (MG) and metabarcoding (MB). Colours represent average of proportion of identifications per sample that were made by both MG and MB (black), MG only (blue), and MB only (red)
In order to check for the reliability of these identifications, we compared the identified genera/species to the checklists of plants for Nee Soon Swamp forest and Singapore (see Methods). Of the 115 genus-level identifications made by metagenomics, 110 were consistent with the Nee Soon Swamp forest list, while two additional ones matched the Singapore checklist and only three were not known for Singapore (Additional file 1: Figure S1a). The corresponding numbers for metabarcoding were as follows: out of 46 identifications, 40 were for plant genera present in the Nee Soon checklist, one was present only in the Singapore checklist and five identifications were present in neither checklist. Overall both methods were reliable at genus level. At species level, both methods had higher mismatches with the Singapore database, as 13.6 % of metagenomics and 25 % of metabarcoding similarities had best matches to extraneous reference sequences. Note, however, that the comparison between metagenomics and metabarcoding at species level is affected by the small numbers of barcodes corresponding to the P6 loop of trnL in the plant database (Additional file 1: Figure S1b).
Congruence of DNA based techniques with field observations
We next tested to what degree the pools of diet species inferred by DNA from the six fecal samples overlapped with the field observations. We first excluded potential misidentifications and synonyms (Additional file 1: Figure S1, yellow/red) and then limited the analyses to genus/family level identifications due to greater uncertainty at species level. Using metagenomics we obtained a set of 53 distinct plant identifications from 33 families. Forty-nine of the 53 identifications were at genus level, while four identifications could be made only to family (Araceae sp., Primulaceae sp., Sapidaceae sp., and Sapotaceae sp. Table 1) and could not be resolved further. Using metabarcoding we obtained 35 distinct plant identifications from 32 families. Twenty-one of the 35 identifications were to genus, while the remaining 14 distinct family level identifications could not be resolved further.
Comparison of these results to diet profile from field studies comprising 27 diet species from 20 families and 24 genera revealed that overall identifications by metagenomics, metabarcoding and field observations corroborated each other, but the DNA based analyses gave larger number of plant identifications. When all three methods were compared, there was high level of congruence for the family level profile with 16 of 20 families of plants from field observations also identified using metagenomics and metabarcoding (Fig. 2a). Due to greater taxonomic resolution achieved by metagenomics, the overlap at genus level was better for metagenomics as compared to metabarcoding (MG: 15/24 genra, MB: 6/24 genera). Lastly, out of the 15 genera observed in HTS based diet analyses and field observations, 11 were found in three or more samples (Table 1). Plants with multiple feeding observations were also present in multiple fecal samples: Fibraurea, Prunus, Hevea, Xanthophyllum, and Litsea were present in six, six, four, four and four samples respectively.

Number of family (a) and genus (b) level identifications using metagenomics, metabarcoding and field observations
Effect of sequencing depth on diet analysis using metagenomics
The completeness of the HTS dietary profile may depend on the sequencing depth. Rarefaction of sequence reads indicated that four of six samples approached an asymptote at sequencing depth of 70–100 million reads while the two most diverse samples (BLM2 and BLM6) showed increasing species diversity at this sequencing depth (Fig. 3). Hence, sequencing ~70 million paired reads (~10 Gbp) would lead to identification of most of the diet items in most samples, although due to the variability in diet across individuals or feeding events, the current sequencing depth may not be sufficient to capture the full dietary breadth of an individual.

Rarefaction curves representing number of plants identified at varying sequencing depths per sample. Rarefaction of plant reads was extrapolated to estimate effect of rarefaction of all reads in the metagenome
Diet of banded leaf monkeys using metagenomics, metabarcoding and field observational data
We built a dietary profile of P. femoralis by combining the above species identifications made from HTS with those from field observations and thus obtained a profile consisting of 38 families. Thirty five of 38 family records could be further resolved to include 60 genera while three family records remained unresolved giving a total of at least 63 plant identifications. We could putatively assign 43 species names to 38 of these genus names (Table 1). They comprise 30 trees, 12 lianas and one shrub. Fibraurea tinctoria (Menispermaceae) and Prunus polystachya (Rosaceae) were found in all six samples, while Xanthophyllum ellipticum, Securidaca philippinensis (Polygalaceae), Hevea (Euphorbiaceae), Bauhinia, Dalbergia (Fabaceae), Litsea (Lauraceae), Strychnos (Loganiaceae), Artocarpus, Ficus (Moraceae), Knema (Myristicaceae) were present in four samples. The dominant families were Fabaceae, Moraceae, Menispermaceae, Rosaceae and Rubiaceae, which were present across all six samples, and Polygalaceae and Lauraceae present in five samples.
Low genetic variability in mitochondrial genomes
A complete mitochondrial genome sequence of 16,548 bp was reconstructed for one sample (BLM5) and used for read map** of the remaining samples. The coverage for the six samples was 10.7–104.7X (HiSeq) and 7.4–41.3X (MiSeq). SNP calling using FreeBayes with ploidy = 1 led to identification of only three variables sites in the mitochondrial genomes (Table 2). Four of the six samples showed polymorphism at position 7791 or 15,572 with good confidence of at least 5× coverage for both alleles. Overall, four different genotypes were recognized separating the individuals for BLM2, BLM4, BLM6 and the three identical samples BLM1, BLM3 and BLM5 (Table 2).
DNA from parasites and other Metazoa in the fecal material
BLAST searches against a parasite SSU rDNA database revealed presence of several protists and nematodes (Table 3). Sequences corresponding to Blastocystis and Entamoeba were present in varying amounts in four and five samples, respectively. Additionally, nematode identifications were made for Strongyloides sp. (3 samples) and Oesophagostomum sp. (one sample). Using the COI database, we recovered sequences mainly corresponding to plants, the primate host and arthropods. Arthropod sequences were found in three samples, and mostly in two (BLM3, BLM6; Additional file 1: Table S7), including Muscidae in both samples, and Sarcophagidae and Drosophilidae (BLM3) and Sepsidae and Lepidoptera (BLM6). At genus level the closest hits were to Dicranosepsis (Sepsidae, BLM6) and Phortica (Drosophilidae, BLM3).
Discussion
Comparison of metagenomics, metabarcoding, and field observational data
We demonstrate the power of metagenomic shotgun sequencing for the characterization of fecal samples and find that it can quickly yield important natural history data for endangered species based on few samples. Using metagenomics we document a diverse diet for the banded leaf monkey comprising at least 53 diet plants from 33 families. There was a good overlap between metagenomics and field observational data, with 15 of 24 genera of observed diet plants found in metagenomics data from merely six samples. Moreover, metagenomics recovered similar number of plants as metabarcoding, as suggested by the comparison of family level profiles whilst providing greater taxonomic resolution by using multiple, longer genetic markers. In addition to a very diverse diet, the shotgun approach also detected previously uncharacterized parasites, and revealed low genetic diversity in mitochondrial genomes of P. femoralis.
There is good agreement between the diet reconstructed based on HTS data and field observations. Nearly half of the plant genera obtained from observational studies (11/24 genera) were also identified in at least three fecal samples. Researchers are more likely to observe feeding events involving frequently utilized diet species and these are also more likely to be present in multiple fecal samples (e.g., Fibraurea, Hevea, Prunus). We thus interpret the good overlap as indirect evidence for the reliability of the diet inferred by metagenomics but note that a dietary profile obtained from six samples is not comprehensive as, e.g., nine of 24 field-observed genera were not detected. Nonetheless, the diet profile obtained with metagenomics was much broader than the profile obtained by field observations. This is not unexpected given that the fieldwork only yielded 31 feeding observations while each fecal sample has the potential to cover ~ 48 h of feeding thus allowing for the identification of rare diet elements. Overall using HTS based methods, our analyses of only six samples added 39 plants to the observational data that had required ~30 months of field work. Field work was still necessary for sample collection, but in the future it can be aided greatly by use of dogs trained to detect feces from target species [10]. However, observational data still has some advantages. Firstly, it can provide information as to which specific individual and which parts of a plant are consumed although the latter can be difficult for food plants in a forest with >700 species of trees and lianas. Secondly, DNA based analyses may not necessarily represent preferred diet plants but also accidental ingestions, such as ingestion of pollen or any other material that may have been associated with the preferred diet items. The latter concern can be overcome by only considering diet items that are found in multiple samples.
When compared to metabarcoding based on one short amplicon, our results are similar to Srivathsan et al. [16] in that the main advantage of metagenomics is higher taxonomic resolution, which can be attributed to utilization of a combination of three barcodes and not limiting the analyses to the P6 loop of trnL. Our results for metabarcoding suggest the PCR-based approaches can amplify nearly all plant families revealed by metagenomics if the primers are universal enough, and thus the trnL approach is useful when a large number of samples have to be multiplexed and a family-level dietary profile is sufficient. To improve on identifications additional genetic markers can be included using methods such as the two-step approach involving group-specific primers proposed by De Barba et al. [18]. Here, the initial family-level identifications were further resolved using amplicons generated by family/taxon-specific nr ITS primers. This is feasible, but would require considerable effort for our samples because trnL sequences from 18 different families could not be assigned to genus/species suggesting that 18 new primer pairs may need to be designed and then used for each sample. Alternatively, metabarcoding could be based on multiplex PCRs using universal primers for multiple short barcodes. However, there is general consensus in the plant barcoding literature that no specific combination of currently used barcodes can be universally applied for species identification [27] and the multiplex PCRs would have to cover multiple markers. A third option may be anchored hybrid enrichment methods which allow for improving representation of multiple regions of interest in a sample [49] under default parameters and the mitochondrial genome of the related P. melalophos (GenBank: NC_008217) as reference. The genome was annotated using MITOS [50] and further manually curated prior to GenBank submission. A single contig was obtained against which reads were mapped back from all six HiSeq and MiSeq datasets using BWA mem [51]. The bam files generated in this process were further filtered to retain only sequences with map** quality of at least 30 and available paired-end reads. We used FreeBayes [52] with ploidy = 1, maximum read mismatch to reference setting at 5 %, minimum coverage for alternate allele at five and variant quality score of at least 30 to identify the variant sites across the six samples. Results obtained using HiSeq and MiSeq datasets from the same sample were first cross checked to ensure there were no differences in the calls for the two runs and then summarised together (see Additional file 1).
Parasites and other eukaryotes
In order to characterize other eukaryotes represented in the fecal samples, reads were matched against COI databases using settings identical to those in the diet analyses. All reads matching the COI database were retrieved and matched to the NT database of GenBank. Identifications were filtered using readsidentifier v1.0 at 95 % and 98 % identities, and only complete overlap between a read and COI sequences was considered. An initial survey of these results revealed matches to mostly plant, primate and insect sequences. Given that we were also interested in identifying potential parasites, we built a target rDNA database of common non-human parasites (Additional file 1: Table S1). SSU rDNA was selected as it has often been used to barcode single-cellular organisms and parasites such as nematodes. Similar to COI analyses, we matched the sequences using MEGABLAST and the retrieved hits were then matched to the NT database to validate the results. The reads were then classified at 98 % similarity and 50 bp overlap using readsidentifier v1.0.
Availability of supporting data
Barcode sequences that matched metagenomics data have been submitted to GenBank with accession numbers KU853075-KU853258. Reference mitochondrial genome has been submitted to GenBank under accession number KU899140. Sequences corresponding to plants from the metagenomic data and the metabarcoding dataset, and plant databases have been archived in LabArchives doi:10.6070/H4000047. COI and parasite databases are available on request. Scripts written specifically for the study are included in the readsidentifier package https://github.com/asrivathsan/readsidentifier.
Abbreviations
- HTS:
-
high throughput sequencing
- MG:
-
metagenomics
- MB:
-
metabarcoding
References
Ceballos G, Ehrlich PR. Mammal population losses and the extinction crisis. Science. 2002;296:904–7.
IUCN. World’s 25 most endangered primates revealed. http://www.iucn.org/?uNewsID=22190. Accessed 27 Nov 2015.
Ceballos G, Ehrlich PR, Barnosky AD, Garcia A, Pringle RM, Palmer TM. Accelerated modern human-induced species losses: entering the sixth mass extinction. Sci Adv. 2015;1:e1400253.
Cracraft J. The urgency of building global capacity for biodiversity science. Biodivers Conserv. 1995;4:463–75.
Tewksbury JJ, Anderson JG, Bakker JD, et al. Natural history’s place in science and society. Bioscience. 2014;64:300–10.
Bradley RD, Bradley LC, Garner HJ, Baker RJ. Assessing the value of natural history collections and addressing issues regarding long-term growth and care. Bioscience. 2014;64:1150–8.
Sitas N, Baillie JEM, Isaac NJB. What are we saving? Develo** a standardized approach for conservation action. Anim Conserv. 2009;12:231–7.
Metrick A, Weitzman ML. Patterns of behaviour in endangered species preservation. Land Econ. 1996;72:1–16.
Ceballos G, Brown JH. Global patterns of mammalian diversity, endemism and endangerment. Conserv Biol. 1995;9:559–68.
Smith DA, Ralls K, Cypher BL. Assessment of scat-detection dog surveys to determine kit fox distribution. Wildl Soc Bull. 2005;33:897–904.
Moreno-Black G. The use of scat samples in primate diet analysis. Primates. 1978;19:215–21.
Kouassi RYW, McGraw SW, Yao PK, et al. Diversity and prevalence of gastrointestinal parasites in seven non-human primates of the Tai National Park, Cote d’Ivoire. Parasite. 2015;22:1.
Kolleck J, Yang M, Zinner D, Roos C. Genetic diversity of endangered Guizhou snub-nosed monkeys (Rhinopithecus brelichi): contrasting results from microsatellite and mitochondrial DNA. PLoS One. 2013;8:e73647.
Bradley BJ, Stiller M, Doran-Sheey DM, Harris T, Chapman CA, Vigilant L, Poinar H. Plant DNA sequences from feces: potential means for assessing diets of wild primates. Am J Primatol. 2007;69:699–705.
Hamad I, Forestier CL, Peeters M, Delaporte E, Raoult D, Bittar F. Wild gorillas as a potential reservoir of Leishmania major. J Infect Dis. 2014;211:267–73.
Srivathsan A, Sha JC, Vogler AP, Meier R. Comparing the effectiveness of metagenomics and metabarcoding for diet analysis of a leaf-feeding monkey (Pygathrix nemaeus). Mol Ecol Resour. 2015;15:250–61.
Quemere E, Hibert F, Miquel C, et al. A DNA metabarcoding study of a primate dietary diversity and plasticity across its entire fragmented range. PLoS One. 2013;8:e58971.
De Barba M, Miquel C, Boyer F, Mercier C, Rioux D, Coissac E, Taberlet P. DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet. Mol Ecol Resour. 2014;14:306–23.
Paula DP, Linard B, Andow DA, Sujii ER, Pires CSS, Vogler AP. Detection and decay rates of prey and prey symbionts in the gut of a predator through metagenomics. Mol Ecol Resour. 2015;15:880–92.
Eisen JA. Environmental shotgun sequencing: its potential and challenged for studying the hidden world of microbes. PLoS Biol. 2007;5:e82.
Ang A, Ismail MRB, Meier R. Reproduction and infant pelage colouration of the banded leaf monkey (Mammalia: Primates: Cercopithecidae) in Singapore. Raffles Bull Zool. 2010;52:411–5.
Ang A, Srivasthan A, Md-Zain BM, Ismail MRB, Meier R. Low genetic variability in the recovering urban banded leaf monkey population of Singapore. Raffles Bull Zool. 2012;60:589–94.
Hüttche C. The ecology, taxonomy and conservation status of the Banded Leaf Monkey (Presbytis femoralis femoralis) in Singapore. Berlin: Free University; 1994.
Nijboer J, Clauss M. The digestive physiology of colobine primates. In: Nijboer J, editor. Fibre intake and faeces quality in leaf-eating primates. Ridderkerk: Ridderprint; 2006.
Hollingsworth PM, Graham SW, Little DP. Choosing and using a plant DNA barcode. PLoS One. 2011;6:e19254.
Wong HF, Tan SY, Koh CY, Siow HJM, Li T, Heyzer A, Ang A, Ismail MRB, Srivathsan A, Tan HTW. Checklist of the plant species of Nee Soon swamp forest, Singapore: Bryophytes to Angiosperms. Singapore: National Parks Board and Lee Kong Chian Natural History Museum; 2013.
Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S. Plant DNA barcoding: from gene to genome. Biol Rev. 2015;90:157–66.
Liu S, Wang X, **e L, Tan M, et al. Mitochondrial capture enriches mito-DNA 100 fold, enabling PCR-free mitogenomic biodiversity analysis. Mol Ecol Resour. 2016;16:470–9.
Baker SC. What to look for in a next generation sequencer. Genet Eng Biotechn N. 2014;34:36–7.
Sheridan C. Illumina claims $1,000 genome win. Nat Biotechnol. 2014;32:115.
Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One. 2015;10:e0128036.
Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012;22:939–46.
Pompanon F, Deagle BE, Symondson WOC, Brown DS, Jarmon SN, Taberlet P. Who is eating what: diet assessment using next generation sequencing. Mol Ecol. 2012;21:1931–50.
Chong KY, Yee ATK, Yeo CK. Biodiversity: linking Singapore’s fragmented habitats. Nature. 2010;465:289.
Kvist L, Martens J, Nazarenko AA, Orell M. Paternal leakage of mitochondrial DNA in the great tit (Parus major). Mol Biol Evol. 2013;20:243–7.
Li M, Schroeder R, Ko A, Stoneking M. Fidelity of capture-enrichment for mtDNA genome sequencing: influence of NUMTs. Nucleic Acids Res. 2012;40:e137.
Karim N, Yang CO. Oesophagostomiasis in man: report of the first Malaysian case with emphasis on its pathology. Malays J Pathol. 1992;14:19–24.
de Melo AL, Mati VLT, Martins WA. Callithrix penicillata as a nonhuman primate model for strongyloidiasis. Primates. 2012;53:303–9.
Arizono N, Yamada M, Tegoshi T, Onishi K. Molecular identification of Oesophagostomum and Trichuris eggs isolated from wild Japanese macaques. Korean J Parasitol. 2012;50:253–7.
Nock CJ, Waters DL, Edwards MA, Bowen SG, Rice N, Cordeiro GM, Henry RJ. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol J. 2011;9:328–33.
Valentini A, Miquel C, Nawaz M, Bellemain E, Coissac E, Pompanon F, Gielly L, Cruaud C, Nascetti G, Wincker P, Swenson JE, Taberlet P. New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Mol Ecol Resour. 2009;9:51–60.
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A, Vermat T, Corthier G, Brochmann C, Willerslev E. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007;35:e14.
Hunt T, Bergsten J, Levkanicova Z, et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science. 2007;318:1913–6.
Ficetola GF, Coissac E, Zundel S, Riaz T, Shehzad W, Bessiere J. Taberlet P Pompanon F An In silico approach for the evaluation of DNA barcodes. BMC Genomics. 2010;11:434.
Bogler AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Huson DH, Weber N. Microbial community analysis using MEGAN. Methods Enzymol. 2013;531:465–85.
Boyer F, Mercier C, Bonin A, Le Bras Y, Taberlet P, Coissac E. Obitools: a unix-inspired software package for DNA metabarcoding. Mol Ecol Resour. 2015. doi:10.1111/1755-0998.12428.
Chong KY, Tan HTW, Corlett RT. A checklist of the total vascular plant flora of Singapore: native, naturalised and cultivated species. Singapore: Raffles Museum of Biodiversity Research; 2009.
Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads- a baiting and iterative map** approach. Nucleic Acids Res. 2013;41:e129.
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. ar**v preprint ar**v:1207.3907 [q-bio.GN]. 2012.
Acknowledgements
We would like to thank the Nee Soon Swamp Forest Survey team and Mirza Rifqi Bin Ismail. Special thanks go to Teo Li Young and Tay Ywee Chieh who were involved in sequencing DNA barcodes for the Nee Soon Swamp forest. We are grateful to Shawn Lum and Chong Kwek Yan for their comments on flora of Singapore. We would like to acknowledge support from Natural History Museum London and Imperial College London HPC division and especially Simon Burbidge and Peter Foster for help with the servers. MiSeq sequencing was funded by Illumina Inc. We would further like to acknowledge the NUS-Imperial college Joint Degree PhD programme. This project was funded by grants R-154-000-539-112 and R-347-000-198-490.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
AS and RM designed the experiments. AS and AA conducted the molecular work. AA conducted the field study and commented on the manuscript. AS conducted the data analyses with advise from RM and APV. AS, APV and RM wrote the manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1:
Supplementary methods, figures and tables. (DOCX 66 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Srivathsan, A., Ang, A., Vogler, A.P. et al. Fecal metagenomics for the simultaneous assessment of diet, parasites, and population genetics of an understudied primate. Front Zool 13, 17 (2016). https://doi.org/10.1186/s12983-016-0150-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12983-016-0150-4