
Abstract
Background
Immunotoxins are antibody-toxin conjugates that bind to surface antigens and exert effective cytotoxic activity after internalization into tumor cells. Immunotoxins exhibit effective cytotoxicity and have been approved by the FDA to treat multiple hematological malignancies, such as hairy cell leukemia and cutaneous T-cell lymphoma. However, most of the internalized immunotoxin is degraded in lysosomes, and only approximately 5% of free toxin escapes into the cytosol to exert cytotoxicity. Many studies have improved immunotoxins by engineering the toxin fragment to reduce immunogenicity or increase stability, but how the antibody fragment contributes to the activity of immunotoxins has not been well demonstrated.
Methods
In the current study, we used 32A9 and 42A1, two anti-GPC3 antibodies with similar antigen-binding capabilities and internalization rates, to construct scFv-mPE24 immunotoxins and evaluated their in vitro and in vivo antitumor activities. Next, the antigen-binding capacity, trafficking, intracellular protein stability and release of free toxin of 32A9 scFv-mPE24 and 42A1 scFv-mPE24 were compared to elucidate their different antitumor activities. Furthermore, we used a lysosome inhibitor to evaluate the degradation behavior of 32A9 scFv-mPE24 and 42A1 scFv-mPE24. Finally, the antigen-binding patterns of 32A9 and 42A1 were compared under neutral and acidic pH conditions.
Results
Although 32A9 and 42A1 had similar antigen binding capacities and internalization rates, 32A9 scFv-mPE24 had superior antitumor activity compared to 42A1 scFv-mPE24. We found that 32A9 scFv-mPE24 exhibited faster degradation and drove efficient free toxin release compared to 42A1 scFv-mPE24. These phenomena were determined by the different degradation behaviors of 32A9 scFv-mPE24 and 42A1 scFv-mPE24 in lysosomes. Moreover, 32A9 was sensitive to the low-pH environment, which made the 32A9 conjugate easily lose antigen binding and undergo degradation in lysosomes, and the free toxin was then efficiently produced to exert cytotoxicity, whereas 42A1 was resistant to the acidic environment, which kept the 42A1 conjugate relatively stable in lysosomes and delayed the release of free toxin.
Conclusions
These results showed that a low pH-sensitive antibody-based immunotoxin degraded faster in lysosomes, caused effective free toxin release, and led to improved cytotoxicity compared to an immunotoxin based on a normal antibody. Our findings suggested that a low pH-sensitive antibody might have an advantage in the design of immunotoxins and other lysosomal degradation-dependent antibody conjugate drugs.
Similar content being viewed by others


Introduction
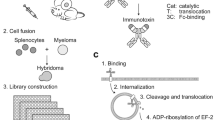
Immunotoxins are chimeric molecules that consist of antibody fragments fused to toxin fragments. Immunotoxins recognize the surface antigen of tumor cells and are internalized with the antigen into tumor cells. Internalized immunotoxins are degraded in lysosomes, and the free toxin fragment is released into the cytosol [1]. The released free toxin fragment of Pseudomonas exotoxin (PE)-type immunotoxin catalyzes the ADP-ribosylation of elongation factor 2, inhibits protein synthesis, and leads to apoptosis [2]. In general, immunotoxins exhibit effective cytotoxicity against tumor cells, which makes them an attractive strategy for cancer therapy [3,4,5,6]. Currently, an immunotoxin against CD22 has achieved exciting success as a treatment for hairy cell leukemia and was approved by the FDA [7], and several immunotoxins targeting solid tumors are also undergoing clinical evaluation [8,9,10,11].
The cytotoxicity of immunotoxins is directly mediated by the free toxin fragment released from lysosomes. However, most immunotoxins are entirely degraded (~ 95%) by a variety of hydrolytic enzymes in acidic lysosomes, and only a small portion of immunotoxins are digested into free toxin that escapes into the cytosol [12]. The details of immunotoxin degradation and free toxin escape into the cytosol remain unclear. Therefore, overcoming the low intracellular utilization of immunotoxins is of great interest in improving the antitumor activity of immunotoxins. Many studies have focused on the engineering of toxin fragments to attenuate immunogenicity [13, 14], reduce off-target toxic effects [15], and extend the half-life of immunotoxins [16, 17]. To date, the optimized PE toxin fragment only maintains the furin cleavage site and domain III, which are important for cytotoxic activity and directly mediate the catalytic reaction of immunotoxins, respectively [18, 19]. Those optimized sequences of the PE fragment are unlikely to be further engineered to improve the intracellular use of immunotoxins without compromising their cytotoxic activity. However, whether antibody fragments can regulate the cytotoxicity of immunotoxins has not been well explored.
In fact, many studies have attempted to develop antibodies with different characteristics to optimize the therapeutic effects of antibody drugs, such as antibodies against different epitopes of HER2, which were reported to internalize with different efficiencies [20]. Additionally, modifying the affinity of antibodies in an acidic environment affected the behavior of the antibody drug in lysosomes. For example, by reducing the affinity of pertuzumab at acidic pH, its related antibody‒drug conjugates (ADCs) showed increased lysosomal delivery and cytotoxicity toward tumor cells [21]. These findings all showed the influence of antibodies on the behavior and functions of antibody conjugates.
Our previous studies established a series of immunotoxins targeting glypican-3 (GPC3) [3], a cell surface antigen that is specifically expressed in hepatocellular carcinoma (HCC) [22]. We selected an antibody targeting the Wnt-binding site of GPC3 to construct a novel anti-GPC3 immunotoxin, which exhibited effective antitumor activity via PE toxin-mediated cytotoxicity and antibody fragment-mediated blocking of cancer signaling [3, 23]. Moreover, we replaced PE38 with mPE24 to deduce the undesirable immunogenicity of the PE toxin and successfully achieved improved antitumor activity by increasing the in vivo safety of immunotoxin [24].
In the current study, we constructed immunotoxins using two anti-GPC3 antibodies with similar affinity and endocytosis efficiency and found that they exhibited significantly different in vitro and in vivo antitumor activities. These two immunotoxins exhibit different lysosomal behaviors, which results in differences in their intracellular protein stabilities and free toxin release. Further investigation indicated that the antigen-binding capacity of these two antibodies varied at acidic pH, which may be the main factor to affect immunotoxin protein stability in the lysosome and the antitumor activity of the immunotoxin.
Methods and materials
Clinical samples
Normal liver tissue and HCC tumor tissue specimens were obtained from 2 male patients who underwent surgical resection for HCC at Jiangsu Taizhou People’s Hospital (Taizhou, Jiangsu) between November 2022 and March 2023. The histologic grade of tumor differentiation was at II, according to the Edmondson grading system. None of the patients had multiple tumor, satellite nodule, vascular invasion or distant metastasis. The patients did not receive anticancer therapies before the surgical operation.
Cell lines
Huh-7 cells were a kind gift from Dr. **n-Wei Wang at the National Cancer Institute (Bethesda, MD, USA). A431, Hep3B and HEK293T cells were purchased from American Type Culture Collection (Manassas, VA, USA). All cells were cultured in DMEM (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (VACCA, St. Louis, MO, USA), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (HyClone, Logan, UT, USA) and were incubated in 5% CO2 at 37 °C. A431 cells were engineered to highly express GPC3 via transfection with a plasmid encoding full-length GPC3. All cell lines were evaluated and validated by their morphology and growth rate. All cell lines were confirmed to be free of mycoplasma contamination using PCR with specific primers.
Plasmids, proteins, and antibodies
The sequence of full-length GPC3 was cloned into the pLVX vector. For protein purification, truncated GPC3 (Q25-S550) was fused to a human Fc tag and cloned into the pFUSE vector. The 32A9 or 42A1 scFv sequence, isolated from the Tomlinson I library to bind human GPC3 [25, 26], was fused to the human Fc tag and cloned into the pFUSE vector. The 32A9 or 42A1 heavy chain variable region and light chain variable region sequences were amplified by adding the IL-2 signal peptide and were inserted into the expression vectors pFUSE-CHIg-HG1 and pFUSE2-CLIg-hk (Invitrogen, San Diego, CA), respectively. All pFUSE plasmids were identified by sequencing and then transfected into 293 T cells. After collecting the supernatant, protein purification was performed using a protein A affinity column (GE Healthcare, Milwaukee, WI, USA). GPC3-His and GPC5-His proteins were purchased from R&D (Minneapolis, MN, USA).
YP7, which is a control mouse monoclonal antibody against GPC3 [27], was used to evaluate GPC3 expression by immunohistochemistry staining, flow cytometry and ELISA.
Structural modeling
The 32A9 or 42A1 scFv model and scFv-mPE24 model was submitted to the protein structure modeling web server Phyre2 [28]. PyMOL was used to analyze and render structural models [29].
ELISA
GPC3-hFc or GPC3-His protein (5 μg/ml) was used to coat ELISA wells at 4 °C overnight. The wells were blocked with PBS buffer containing 3% milk and 0.05% Tween-20 for 0.5 h at 37 °C. The antibodies or phages were added to the wells and incubated at 37 °C for 0.5 h. After washing with PBS (containing 0.05% Tween-20) buffer for 3 times, goat anti-human Fc, kappa chain HRP antibody (for 32A9 or 42A1 IgG) (Life Tech, Peoria, IL, USA) or rabbit anti-M13 HRP antibody (for phage) (GE Healthcare, Milwaukee, WI, USA) was added to the wells and incubated at 37 °C for 0.5 h. TMB and sulfuric acid were added to detect the OD450nm value.
Western blotting
The cells were lysed with RIPA/PMSF/protease inhibitor buffer on ice and centrifuged to extract the protein. For the non-reduced analysis, the native protein is treated with SDS prior to separation to mask the protein native charges. For reduced analysis, the sample is treated with SDS and dithiothreitol (DDT) to reduce the native protein structure. Then the total proteins were separated on SDS-PAGE and transferred onto the PVDF membrane (Merck Millipore, Tullagreen, Carrigtwohill, Ireland). After blocking with 5% milk overnight, the PVDF membranes were incubated with primary antibodies for 1 h at room temperature. Anti-β-actin HRP (Sigma, St. Louis, MO, USA) was used as a loading control. The ImageJ software was used to quantify the gray value of the protein bands.
Flow cytometry
Cells were trypsinized into single cell suspensions and being fully dispersed by a pipette after centrifugation and were counted using a cell counting plate. 5 × 105 A431-GPC3 cells were incubated with 5 µg/ml of the indicated antibodies or different concentrations of immunotoxins for 1 h on ice and then incubated with a 1:200 dilution of anti-human PE antibody (Thermo, Pudong New Area, Shanghai) or rabbit anti-Pseudomonas exotoxin A antibody (Sigma, St. Louis, MO, USA) and goat anti-rabbit PE (Abcam, Cambridge, England) for 1 h on ice. After washing with PBS buffer, unstained cells and labeled cells with second fluorescence antibody only were defined as negative cell group, then the positive cell group was gated and continuously collected to 1 × 104 cells using FACS Calibur (BD Biosciences, San Jose, CA, USA) for further analysis by using FlowJo 7.6.
Antibody internalization detection
A total of 3 × 105 A431-GPC3 cells were seeded into 24-well plates and cultured overnight at 80–90% density. A431-GPC3 cells were coincubated with 50 µg/ml Alexa 488-labeled 32A9 IgG or Alexa 488-labeled 42A1 IgG immediately (0), 0.5, 1, 2, and 4 h at 37 °C. A431-GPC3 cells were stripped with 0.2 M glycine buffer (pH 2.5) after washing with PBS buffer. The digested cell suspensions were analyzed using FACS Calibur (BD Biosciences, San Jose, CA, USA).
Immunotoxin purification
The sequence of 32A9 or 42A1 scFv was fused to a truncated and deimmunized Pseudomonas exotoxin (mPE24) fragment and then cloned into the pMH212 vector [13]. 100 uL of BL21 competent cells (Weidi Biotechnology, Minhang District, Shanghai) were transfected with the plasmid and induced with 1 mM IPTG for 1.5 h at 37 °C. The inclusion body was collected, washed, and lysed using lysozymes to obtain the raw extraction of the recombinant protein. The recombinant protein was denatured and refolded, and the buffer was exchanged via overnight dialysis. The refolded recombinant protein was acquired using chromatography with ÄKTA pure (GE Healthcare, Milwaukee, WI, USA): we used Q-Sepharose and Mono-Q (GE Healthcare, Milwaukee, WI, USA) ion-exchange chromatography for separating molecules on the basis of charge, and finally eluted immunotoxin protein into PBS buffer using a TSK (Tosoh, Changning, Shanghai) gel-filtration step that separated molecules on the basis of size, and determined protein concentration, aliquot, and freeze at 70 °C.
Co-localization analysis
A total of 5 × 104 A431-GPC3 cells were seeded into confocal dish and cultured overnight at 70–80% density. After incubating A431-GPC3 cells with 50 µg/ml immunotoxin on ice for 1 h, the cells were washed with PBS and labeled with anti-Pseudomonas exotoxin A antibody (Sigma, St. Louis, MO, USA) and Alexa647-conjugated goat anti-rabbit (Invitrogen, San Diego, CA, USA) on ice for 1 h. The treated cells were coincubated with DMEM immediately (0), 1 and 3 h at 37 °C to internalize labeled immunotoxins. Thereafter, the cells were incubated with lysosome tracer (Beyotime, Songjiang, Shanghai) for 1 min and DAPI (Beyotime, Songjiang, Shanghai) in the dark for 1 min and observed under laser confocal microscope in randomly selected fields.
Antibody and immunotoxin stability detection
A total of 3 × 105 A431-GPC3 cells were seeded into 24-well plates and cultured overnight at 80–90% density. After incubating A431-GPC3 cells with 50 μg/ml immunotoxin at 37 °C for 1 h, the cells were washed with PBS and 0.2 M glycine buffer (pH 2.5). The treated cells were coincubated with DMEM immediately (0), 0.5, 1, 2, and 4 h at 37 °C to degrade accumulated immunotoxin. A431-GPC3 cells were washed and lysed for Western blot analysis. The antibodies used included anti-Pseudomonas exotoxin A antibody (Sigma, St. Louis, MO, USA), anti-rabbit HRP (Jackson Laboratory, Bar Harbor, ME, USA) and anti-β-actin HRP (Sigma, St. Louis, MO, USA). A431 cells were also treated as negative control.
To block lysosomal degradation, 50 µM chloroquine (MCE, St. Monmouth Junction, NJ, USA) was added to the wells for 12 h before incubating the cells with immunotoxin during immunotoxin degradation.
Free toxin release detection
A total of 3 × 105 A431-GPC3 cells were seeded into 24-well plates and cultured overnight at 80–90% density. A431-GPC3 cells were coincubated with 50 µg/ml immunotoxin immediately (0), 0.5, 1, 2, 4, and 8 h at 37 °C to accumulate free toxin. A431-GPC3 cells were stripped with 0.2 M glycine buffer (pH 2.5) and lysed for Western blot analysis. The antibodies used included anti-Pseudomonas exotoxin A antibody (Sigma, St. Louis, MO, USA), anti-rabbit HRP (Jackson Laboratory, Bar Harbor, ME, USA) and anti-β-actin HRP (Sigma, St. Louis, MO, USA). A431 cells were also treated as a negative control.
To block lysosomal degradation, 50 µM chloroquine (MCE, St. Monmouth Junction, NJ, USA) was added to the wells for 12 h before incubating cells with immunotoxin during immunotoxin degradation.
Cytotoxicity of immunotoxins
A total of 1 × 104 cells were seeded into 96-well plates and cultured overnight at 80–90% density. Different concentrations of immunotoxin were added to the wells. After 72 h, CCK8 (Beyotime, Songjiang, Shanghai) is added to each well, and the incubation is carried out for 1 h at 37 °C. The absorbance of the sample at 450 nm is measured. Cytotoxicity is expressed as 50% inhibition of cell viability, which is halfway between the level of viability in the absence of toxin and that in the presence of 10 mg/ml of cycloheximide.
Animal tests
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Nan**g Medical University under the ethical review number “No. IACUC-2107004”. 60 four- to six-week-old female BALB/c nude mices (BALB/cJGpt-Foxn1nu/Gpt) were purchased from GemPharmatech (Guangzhou, China). To evaluate the antitumor activity of the immunotoxins, 5 × 106 A431-GPC3 cells were subcutaneously inoculated into five-week-old nude mice. When the average tumor size reached ~ 100 mm3, according to the order of tumor size, mice bear moderate tumor were selected for immunotoxin treatment, n = 5/group. The mice were treated daily with 2.5 or 5 mg/kg immunotoxin via i.v. injection. Tumor dimensions were determined using calipers, and tumor volume was calculated using the formula V = 1/2 ab2, where a and b represent tumor length and width, respectively.
Statistical analysis
Representative results were obtained from at least three independent experiments. All group data (except otherwise indicated) are expressed as the means ± standard deviations (SD) of a representative experiment performed at least in triplicate, and similar results were obtained in at least 3 independent experiments. All statistical analyses were performed using GraphPad Prism 5.0. Two-tailed Student’s t tests and paired Student’s t test were used for statistical analyses, and *P < 0.05 indicated significance.
Results
Immunotoxin against GPC3 effectively induced cytotoxicity in HCC cells
HCC is the major subtype of primary liver cancer, but it lacks effective treatment. GPC3 is an oncofetal antigen that is specifically expressed in HCC patients [22, 30]. Analysis of the TCGA database and clinical samples of HCC patients revealed that GPC3 was abnormally expressed in HCC patients but not in normal liver tissue (Table 1; Fig. 1A, B). GPC3 exhibited strong membrane expression in HCC cells, which makes it a potent target for immunotherapy (Fig. 1C). Due to its rapid endocytosis, GPC3 is also a suitable target for antibody conjugates [3, 31]. HN3-mPE24, which is the immunotoxin against GPC3 developed in our previous study [24], effectively killed GPC3-positive HCC cells (Fig. 1D). Therefore, we took advantage of the anti-GPC3 immunotoxin to investigate the regulatory effect of the antibody on the function of the immunotoxin.

GPC3 is a potent target for develo** immunotoxin strategies in liver cancer. A The transcriptional expression of GPC3 in liver cancer from The Cancer Genome Atlas Program (TCGA database). n = 369 in the LIHC (liver hepatocellular carcinoma) group, n = 50 in the normal liver tissue group, Values represent the mean ± SD, ***P < 0.0001 (two-tailed Student’s t-test). B Immunohistochemical staining of GPC3 in HCC patient tumor tissues and normal liver tissues. C Flow cytometry was used to detect the cell surface expression of GPC3 in HCC cells. A431 cells and A431-GPC3 cells are shown as negative and positive controls, respectively. D WST assay to detect the cytotoxicity of the anti-GPC3 immunotoxin (HN3-mPE24). Values represent the mean ± SD, n = 3 individual tests, with ***P < 0.001 (two-tailed Student’s t-test)
32A9 scFv-mPE24 has more effective antitumor activity than 42A1 scFv-mPE24 in vitro and in vivo
We isolated two human antibodies that target GPC3, 32A9 and 42A1, by phage display in our previous work [25, 26] (Fig. 2A). The two antibodies showed similar binding activity for cell surface GPC3 (Fig. 2B) and triggered similar endocytosis when they targeted the antigen (Fig. 2C). These results indicated that both 32A9 and 42A1 can be used to construct anti-GPC3 antibody conjugates. We fused the single-chain fragment of the variable region of 32A9 and 42A1 to mPE24, a deimmunized PE fragment [42,43,44], be captured by the neonatal Fc receptor (FcRn) and be recycled into the extracellular space, which improved the pharmacokinetics or pharmacodynamics of antibodies [45]. Moreover, pH-sensitive binding also enhanced the recycling rate of certain antigens [41]. A recent study found that ipilimumab, which is a clinically used antibody against CTLA-4 [46], markedly downregulated the cell surface expression level of CTLA-4 by disrupting CTLA-4 recycling, and the use of an antibody with pH-sensitive binding or increasing the pH sensitivity by introducing designed tyrosine-to-histidine mutations prevented antibody-triggered lysosomal CTLA-4 downregulation. Furthermore, this strategy also more effectively depleted tumor-infiltrating Tregs, which provided safer and more effective immunotherapy [47,48,49,50]. Many studies have developed antibodies with better binding affinity under acidic pH conditions than neutral pH conditions [40, 41]. These antibodies may induce fewer off-tumor effects and improve the safety of immunotherapy by taking advantage of the acidic tumor microenvironment. For example, an antibody with pH-responsive HER2 targeting had superior affinity and inhibited tumor spheroid growth more efficiently under acidic conditions [51]. Similarly, pH-responsive binding-based CAR-T cells function only within the acidic tumor microenvironment in a pH-dependent manner [52].
We isolated two distinct antibodies with similar antibody properties that showed different responsiveness to the target antigen under low pH conditions. We proved that the pH-responsive binding property of the antibody would primarily determine the intracellular stability and free toxin release of the immunotoxins. However, a more serious evaluation is needed to exclude whether the different intracellular behaviors of immunotoxins are epitope dependent, although two antibodies showed similar internalization rates after binding GPC3. Both 32A9 and 42A1 antibodies were isolated from the Tomlinson I phage library [53]. Due to the sequence characteristics of this library, the specific recognition of antigen was mainly determined by the heavy chain CDR2 and light chain CDR3 of antibody. In theory, the interactions effected by the change of pH contains salt bridges and hydrogen bonding in protein folding and protein–protein interaction [35]. Comparing the heavy chain CDR2 and light chain CDR3 of the two antibodies, we find that the light chain CDR3 of 32A9 has the sequence DYAYPY, whereas 42A1 has STSYPT. In this case, only 32A9 has an aspartate where 42A1 has uncharged residues. This difference might mediate the resistance of 42A1 to acidic pH when binding to GPC3. Based on these clues, we are considering to perform single-residue histidine scanning mutagenesis [54, 55] to obtain engineered 32A9 mutant variants with less pH sensitivity or 42A1 mutant variants with improved pH sensitivity for binding GPC3. The mutant variants, in parallel with their parental antibody, will be further evaluated in immunotoxin format because they all target the same epitope. Considering that the pH response differences of 32A9 and 42A1 were also possibly determined by the ionizable groups locating on the antigen epitope region, we would perform detailed analysis on each side and combined with histidine scanning mutagenesis in the future.
The liver is the major metabolic organ of the human body, and its detoxifying features generally cause resistance to chemo-drug treatment [56]. Therefore, immunotoxins may provide a feasible strategy for treating liver cancer because they cause cytotoxic killing via an enzymatic reaction to inhibit tumor cell protein synthesis as a recombinant protein drug instead of chemical drugs. Our findings provide new insight for designing effective immunotoxins and are meaningful for promoting the development of liver cancer therapy.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- LIHC:
-
Liver hepatocellular carcinoma
- GPC3:
-
Glypican-3
- scFv:
-
single-chain fragment of the variable region
- PE:
-
Pseudomonas exotoxin A
- ADCs:
-
Antibody-drug conjugates
- FcRn:
-
Neonatal Fc receptor
References
Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–65.
Akbari B, Farajnia S, Khosroshahi SA, Safari F, Yousefi M, Dariushnejad H, et al. Immunotoxins in cancer therapy: review and update. Int Rev Immunol. 2017;36:207–19.
Gao W, Tang ZW, Zhang YF, Feng MQ, Qian M, Dimitrov DS, et al. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat Commun. 2015;6:6536.
Schwemmlein M, Stieglmaier J, Kellner C, Peipp M, Saul D, Oduncu F, et al. A CD19-specific single-chain immunotoxin mediates potent apoptosis of B-lineage leukemic cells. Leukemia. 2007;21:1405–12.
Hamlin PA, Fanale MA, Park SI, Valacer DJ, Higgins J, Younes A, et al. Data from the first CD20-targeted immunotoxin, MT-3724, in a phase I/Ib study in relapsed/refractory (R/R) non-Hodgkin’s B-cell lymphoma (NHL). Blood. 2016;128:4200–4200.
Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–90.
Tong JTW, Harris PWR, Brimble MA, Kavianinia I. An insight into FDA approved antibody-drug conjugates for cancer therapy. Molecules. 2021;26:5847.
Kreitman RJ, Hassan R, FitzGerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274–9.
Li M, Liu ZS, Liu XL, Hui Q, Lu SY, Qu LL, et al. Clinical targeting recombinant immunotoxins for cancer therapy. Onco Targets Ther. 2017;10:3645–65.
Pai LH, Bookman MA, Ozols RF, Young RC, Smith JW, Longo DL, et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J Clin Oncol. 1991;9:2095–103.
Minckwitz GV, Harder S, Hövelmann S, Jäger E, Al-Batran SE, Loibl S, et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005;7:R617–26.
Ogata M, Chaudhary VK, Pastan I, FitzGerald DJ. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J Biol Chem. 1990;265:20678–85.
Liu WH, Onda M, Lee B, Kreitman RJ, Hassan R, **ang LM, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci USA. 2012;109:11782–7.
Mazor R, Eberle JA, Hu XB, Vassall AN, Onda M, Beers R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci USA. 2014;111:8571–6.
Wang J, Han L, Chen JS, **e YQ, Jiang H, Zhu JW. Reduction of non-specific toxicity of immunotoxin by intein mediated reconstitution on target cells. Int Immunopharmacol. 2019;66:288–95.
Fleming BD, Urban DJ, Hall MD, Longerich T, Greten TF, Pastan I, et al. Engineered anti-GPC3 immunotoxin, HN3-ABD-T20, produces regression in mouse liver cancer xenografts through prolonged serum retention. Hepatology. 2020;71:1696–711.
Guo R, Guo WJ, Cao L, Liu H, Liu JY, Xu H, et al. Fusion of an albumin-binding domain extends the half-life of immunotoxins. Int J Pharm. 2016;511:538–49.
Weldon JE, **ang LM, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113:3792–800.
Hollevoet K, Mason-Osann E, Liu XF, Imhof-Jung S, Niederfellner G, Pastan I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. 2014;13:2040–9.
Cheng J, Liang M, Carvalho MF, Tigue N, Faggioni R, Roskos LK, et al. Molecular mechanism of HER2 rapid internalization and redirected trafficking induced by anti-HER2 biparatopic antibody. Antibodies (Basel). 2020;9(3):49.
Kang JC, Sun W, Khare P, Karimi M, Wang X, Shen Y, et al. Engineering a HER2-specific antibody-drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat Biotechnol. 2019;37(5):523–6.
Li N, Gao W, Zhang YF, Ho M. Glypicans as cancer therapeutic targets. Trends Cancer. 2018;4:741–54.
Feng MQ, Gao W, Wang RQ, Chen WZ, Man YG, Figg WD, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2013;110:E1083–91.
Wang C, Gao W, Feng M, Pastan I, Ho M. Construction of an immunotoxin, HN3-mPE24, targeting glypican-3 for liver cancer therapy. Oncotarget. 2016;8:20.
Liu X, Gao F, Jiang L, Jia M, Ao L, Lu M, et al. 32A9, a novel human antibody for designing an immunotoxin and CAR-T cells against glypican-3 in hepatocellular carcinoma. J Transl Med. 2020;18(1):295.
Ye W, Liu X, He R, Gou L, Lu M, Yang G, et al. Improving antibody affinity through in vitro mutagenesis in complementarity determining regions. J Biomed Res. 2022;36(3):155–66.
Phung Y, Gao W, Man Y-G, Nagata S & Ho M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs. 2012;4:592–599.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58.
Rigsby RE, Parker AB. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem Mol Biol Educ. 2016;44:433–7.
Ho M, Kim H. Glypican-3: a new target for cancer immunotherapy. Eur J Cancer. 2011;47:333–8.
Capurro MI, Shi W, Filmus J. LRP1 mediates Hedgehog-induced endocytosis of the GPC3–Hedgehog complex. J Cell Sci. 2012;125:3380–9.
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–66.
Zhang X, Lin Y, Gillies RJ. Tumor pH and its measurement. J J Nuclear Med. 2010;51:1167–70.
Zeng J, Shirihai OS, Grinstaff MW. Modulating lysosomal pH: a molecular and nanoscale materials design perspective. J Life Sci. 2020;2:25–37.
Dumetz AC, Chockla AM, Kaler EW, Lenhoff AM. Effects of pH on protein-protein interactions and implications for protein phase behavior. Biochim Biophys Acta. 2008;1784(4):600–10.
May KL, Yan Q, Tumer NE. Targeting ricin to the ribosome. Toxicon. 2013;69:143–51.
Singh RC, Alam A, Singh V. Role of positive charge of lysine residue on ribosome-inactivating property of gelonin. Indian J Biochem Biophys. 2001;38(5):309–12.
Sandvig K, Skotland T, van Deurs B, Klokk TI. Retrograde transport of protein toxins through the Golgi apparatus. Histochem Cell Biol. 2013;140:317–26.
Baluna R, Ghetie V, Oppenheimer-Marks N, Vitetta ES. Fibronectin inhibits the cytotoxic effect of ricin A chain on endothelial cells. Int J Immunopharmacol. 1996;18(6–7):355–61.
Igawa T, Mimoto F, Hattori K. pH-dependent antigen-binding antibodies as a novel therapeutic modality. Biochim Biophys Acta. 2014;1844:1943–50.
Klaus T, Deshmukh S. pH-responsive antibodies for therapeutic applications. J Biomed Sci. 2021;28:11.
Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Tadamitsu Kishimoto T, et al. Resha** a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993;53:851–6.
Chaparro-Riggers J, Liang H, DeVay RM, Bai LF, Sutton JE, Chen W, et al. Increasing serum half-life and extending cholesterol lowering by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem. 2012;287:11090–7.
Fukuzawa T, Sampei Z, Haraya K, Ruike Y, Shida-Kawazoe M, Yuichiro Shimizu Y, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep. 2017;7:1080.
Delidakis G, Kim JE, George K, Georgiou G. Improving antibody therapeutics by manipulating the fc domain: immunological and structural considerations. Annu Rev Biomed Eng. 2022;24:249–74.
Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov. 2022;21:509–28.
Liu Y, Zheng P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci. 2020;41:4–12.
Altman A, Kong KF. pH-sensitive anti-CTLA4 antibodies: yes to efficacy, no to toxicity. Cell Res. 2019;29:601–2.
Gao H, Cai HY, Liu J, Wang XX, Zheng P, Devenport M, et al. Structure of CTLA-4 complexed with a pH-sensitive cancer immunotherapeutic antibody. Cell Discov. 2020;6:79.
Zhang Y, Du XX, Liu MY, Tang F, Zhang P, Ai CX, et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. 2019;29:609–27.
Sulea T, Rohani N, Baardsnes J, Corbeil CR, Deprez C, Cepero-Donates Y, et al. Structure-based engineering of pH-dependent antibody binding for selective targeting of solid-tumor microenvironment. mAbs. 2020;12:1682866.
Short JM, Chang HW, Frey G. Conditionally active chimeric antigen receptors for modified t-cells. US-2019010220-A1[P] 2019. https://patents.google.com/patent/US20190010220A1.
De Wildt RM, Mundy CR, Gorick BD, Tomlinson IM. Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat Biotechnol. 2000;18(9):989–94.
Dumetz AC, Chockla AM, Kaler EW, Lenhoff AM. Effects of pH on protein-protein interactions and implications for protein phase behavior. Biochim Biophys Acta. 2008;1784(4):600–10.
Zou WJ, Huang CC, Sun Q, Zhao KL, Gao HY, Su R, et al. A stepwise mutagenesis approach using histidine and acidic amino acid to engineer highly pH-dependent protein switches. 3 Biotech. 2022;12:21.
Bao MH, Wong CC. Hypoxia, metabolic reprogramming, and drug resistance in liver cancer. Cells. 2021;10:1715.
Acknowledgements
We thank our colleagues Dr. Yonglin Yang (Taizhou People's Hospital) for providing HCC patient samples and Mitchell Ho (National Cancer Institute; NCI) for isolating the YP7 antibody.
Funding
This research was supported by the National Natural Science Foundation of China (Nos. 81972284, 81773260 and 82273239) and the National Natural Science Foundation Youth Project of Jiangsu, China (No. BK20171047).
Author information
Authors and Affiliations
Contributions
X.L., Q.T., J.W., X.W., G.Y., Y.L., M.L. and W.Y. performed the experiments. W.G., X.L., A.S., T.D., L.S., F.L. and M.Z. analyzed the data. W.G. and T.J. designed and performed the research. W.G. and X.L. wrote the manuscript, and W.G., T.D., L.S. F.L. and S.M. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
For clinical samples, the Ethical Committee of Jiangsu Taizhou People’s Hospital approved this study (IRB ID: KY-2022-172-01), and each patient provided written informed consent. For animal tests, all mice received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. All mice were treated under the protocol approved by the Ethics Committee of the Animal Core Facility of Nan**g Medical University. "Regulation mechanism of pH responsive antibody for anti-tumor activity of antibody conjugate" has been approved by the Institutional Animal Care and Use Committee (IACUC) under the ethical review number "No. IACUC-2107004". The research project, funded by the Natural Science Foundation of China (NSFC) under the grant numbers 81972284/81773260, is conducted in compliance with relevant ethical and legal guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest with the contents of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, X., Tan, Q., Wen, J. et al. Improving the cytotoxicity of immunotoxins by reducing the affinity of the antibody in acidic pH. J Transl Med 21, 572 (2023). https://doi.org/10.1186/s12967-023-04210-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04210-7