
Abstract
Background
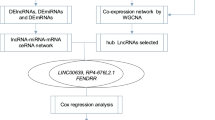
Lung adenocarcinoma (LUAD) is the most common subtype of nonsmall-cell lung cancer (NSCLC) and has a high incidence rate and mortality. The survival of LUAD patients has increased with the development of targeted therapeutics, but the prognosis of these patients is still poor. Long noncoding RNAs (lncRNAs) play an important role in the occurrence and development of LUAD. The purpose of this study was to identify novel abnormally regulated lncRNA–microRNA (miRNA)–messenger RNA (mRNA) competing endogenous RNA (ceRNA) networks that may suggest new therapeutic targets for LUAD or relate to LUAD prognosis.
Methods
We used the SBC human ceRNA array V1.0 to screen for differentially expressed (DE) lncRNAs and mRNAs in four paired LUAD samples. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed to annotate the DE lncRNAs and mRNAs. R bioinformatics packages, The Cancer Genome Atlas (TCGA) LUAD database, and Kaplan–Meier (KM) survival analysis tools were used to validate the microarray data and construct the lncRNA–miRNA–mRNA ceRNA regulatory network. Then, quantitative real-time PCR (qRT-PCR) was used to validate the DE lncRNAs in 7 LUAD cell lines.
Results
A total of 2819 DE lncRNAs and 2396 DE mRNAs (P < 0.05 and fold change ≥ 2 or ≤ 0.5) were identified in four paired LUAD tissue samples. In total, 255 of the DE lncRNAs were also identified in TCGA. The GO and KEGG analysis results suggested that the DE genes were most enriched in angiogenesis and cell proliferation, and were closely related to human cancers. Moreover, the differential expression of ENST00000609697, ENST00000602992, and NR_024321 was consistent with the microarray data, as determined by qRT-PCR validation in 7 LUAD cell lines; however, only ENST00000609697 was associated with the overall survival of LUAD patients (log-rank P = 0.029). Finally, through analysis of ENST00000609697 target genes, we identified the ENST00000609697–hsa-miR-6791-5p–RASL12 ceRNA network, which may play a tumor-suppressive role in LUAD.
Conclusion
ENST00000609697 was abnormally expressed in LUAD. Furthermore, downregulation of ENST00000609697 and its target gene RASL12 was associated with poor prognosis in LUAD. The ENST00000609697–hsa-miR-6791-5p–RASL12 axis may play a tumor-suppressive role. These results suggest new potential prognostic and therapeutic biomarkers for LUAD.
Similar content being viewed by others


Introduction
Lung cancer is the second most commonly diagnosed cancer worldwide, with 2.21 million new cases annually, and is the most common cause of cancer death (1.79 million deaths annually) [1]. Approximately 85% of lung cancer cases are nonsmall-cell lung cancer (NSCLC), and lung adenocarcinoma (LUAD) is currently the most common histological subtype of NSCLC [2,3,4]. Patients with late-stage disease at diagnosis have a poor prognosis. However, the occurrence, development, and prognosis of tumors are not only related to pathological type and clinical stage, but also closely related to abnormal gene expression in tumor cells [5] Early prevention and continuous improvements in targeted drugs support the clinical translation of the lung cancer treatment model, prolong the progression-free survival (PFS) and overall survival (OS) of patients, and improve their prognosis [6]. However, the prognosis of LUAD is still poor due to local recurrence or distant metastasis. Therefore, there is an urgent need to predict and explore more biomarkers for early diagnosis and therapeutic targets in LUAD.
Long noncoding RNAs (lncRNAs) are noncoding RNA (ncRNA) molecules of more than 200 nucleotides that are most commonly not translated into protein, but are crucial players in diverse cellular and physiological functions [7, 8]. Recently, with the development and application of high-throughput sequencing and gene chip technologies, researchers have found that lncRNAs play important roles in the occurrence and development of a variety of tumors [9]. LncRNAs that are abnormally expressed in tumor tissues can not only be used as specific tumor biomarkers for early diagnosis and prognosis, but also directly interact with DNA, messenger RNA (mRNA), or protein to regulate chromatin modification or structure or to affect gene transcription, splicing, and translation [10]. In general, lncRNAs can regulate a variety of physiological and pathological processes in tumor development, such as cell proliferation, differentiation, migration, and invasion; stem cell reprogramming; tumorigenesis; and drug resistance [11,12,13,14,15]. Competing endogenous RNAs (ceRNAs) are RNAs containing microRNA (miRNA) recognition elements (MREs) [16] that can regulate the expression of genes harboring the corresponding MRE or the expression of proteins by competitively binding to miRNAs [17]. LncRNAs can also act as ceRNAs and function within lncRNA–miRNA–mRNA ceRNA networks [17, 18]. Perturbation of ceRNA networks may affect diseases and explain disease processes; thereby, presenting opportunities for new therapies [17]. For instance, multiple studies have demonstrated that patients with various cancers with high HOTAIR expression exhibit higher lymphatic invasion and shorter survival times [19,20,1: 255 overlap** genes).
Annotation analyses of the DE lncRNAs and mRNAs
GO analysis was used to annotate gene functions and standardize the descriptions of the DE genes according to the biological process (BP), cellular component (CC), and molecular function (MF) categories. We analyzed the cis-regulated lncRNAs and found that most of the top 30 GO terms enriched by the upregulated and downregulated genes (i.e., DE lncRNAs and mRNAs) were in the BP and CC categories (Fig. 3A, B). The top three descriptive terms enriched by the DE lncRNAs were atomic septum development, structural molecule activity conferring elasticity, and embryonic digestive tract morphogenesis (Fig. 3C). However, condensed chromosome outer kinetochore, cell migration involved in heart development, and regulation of vasculogenesis were the top three descriptive terms enriched by the DE mRNAs (Fig. 3D). Moreover, all the DE lncRNAs and mRNAs were involved in angiogenesis and cell proliferation.

GO and KEGG enrichment analysis of DE lncRNAs and mRNAs. Barplot of the top 30 enriched GO classification terms for DE lncRNAs (A) and DE mRNAs (B). Bubble plot of the top 30 GO level 2 terms enriched by the DE lncRNAs (C) and DE mRNAs (D). Bubble plot of the top 30 KEGG pathways enriched by the DE lncRNAs (E) and DE mRNAs (F). G, H Barplot of the top 30 KEGG classifications for the DE lncRNAs (G) and DE mRNAs (H). GeneRatio ≥ 2, P < 0.05
The KEGG database, which enables pathway analysis of DE genes to identify biological functions, is divided into the following six classifications: cellular processing, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems. Comprehensive analysis of the KEGG classification results for the DE lncRNAs (Fig. 3G) and DE mRNAs (Fig. 3H) showed enrichment mainly in the terms signal transduction, immune system, and cancer: overview. Moreover, KEGG pathway enrichment analysis suggested that the DE genes were enriched mainly in vascular smooth muscle contraction, focal adhesion, and the TGF beta signaling pathway (Fig. 3E, F). Further analysis of the KEGG pathway term human diseases showed that these DE genes were closely related to small-cell lung cancer, NSCLC, melanoma, glioma, prostate cancer, thyroid cancer, colorectal cancer (CRC), and other tumors (Fig. 3G, H).
Analysis of lncRNA target genes
To further clarify the functional annotations of the DE genes, we determined the intersection of the target genes of the 1302 DE lncRNAs and the 2396 DE mRNAs, and the resulting 523 common DE genes were selected with Venn diagram software (Fig. 4A). GO analysis showed that these genes were also enriched in the CC and BP categories. Moreover, the main enriched terms were extracellular matrix, myosin complex, and cytoskeleton in the CC category (Fig. 4B); signal transduction in the BP category (Fig. 4C); and peptidase activity in the MF category (Fig. 4D). The KEGG analysis results showed that these genes were enriched mainly in the pathways focal adhesion, axon guidance, differentiated cardiomyopathy, and melanoma (Fig. 4E). These results suggested that the identified DE genes may play important roles in cell morphology, adhesion, intercellular connections, and signal transduction.

GO and KEGG enrichment analysis of the intersection between DE lncRNA target genes and DE mRNAs. A Venn diagram of DE mRNAs and DE lncRNA target genes. B Cell component (CC). C Biological process (BP). D Molecular function (MF). E KEGG pathway. The y‐axis shows significantly enriched categories for the targets, and the x axis shows the enrichment scores of these terms. The bar plot height indicates the number of genes in the functional area
Candidate DE lncRNA validation in LUAD cell lines and OS analysis
We selected four candidate DE lncRNAs from the 255 overlap** genes: two downregulated genes (ENST00000609697 and ENST00000443224) and two upregulated genes (ENST00000602992 and NR_024321). To confirm the screening results, the expression of the 4 DE lncRNAs was validated in 7 LUAD cell lines and compared with that in the BEAS-2B cell line using qRT-PCR (Fig. 5A). The expression of ENST00000609697 and ENST00000443224 showed a significant decreasing trend in almost all the LUAD cell lines, consistent with the microarray data (P < 0.05), while ENST00000443224 was upregulated in H1993 cells (P < 0.05) (Fig. 5A). The significant increasing trend (P < 0.05) in ENST00000602992 and NR_024321 expression was also consistent with the microarray data, but the increasing trend in NR_024321 expression was not obvious in H2228 cells (Fig. 5B). Next, we downloaded gene expression data and patient follow‐up data from the TCGA dataset to elucidate whether these candidate genes are potential prognostic markers for LUAD. Through TCGA dataset analysis, we found that ENST00000609697 was downregulated (P < 0.001) (Fig. 5C) and was the only candidate gene related to the prognosis of LUAD (log-rank P = 0.029) (Fig. 5D). ENST00000602992 and NR_024321 were upregulated in the TCGA dataset (P < 0.001) (Additional file 2: Fig. S1A, B). However, ENST00000602992 was not associated with the prognosis of LUAD (P = 0.24) (Additional file 2: Fig. S1C), and NR_024321 upregulation was not positively correlated with good prognosis in LUAD (P = 0.018) (Additional file 2: Fig. S1D). However, the downregulation of ENST00000609697 was positively correlated with good prognosis in LUAD; and therefore, this gene was considered a candidate biomarker that may function as a tumor suppressor.

Validation of candidate lncRNAs. A The expression of the downregulation candidate lncRNAs in 7 LUAD cell lines and BEAS-2B cells was determined by qRT-PCR. B The expression of upregulation candidate lncRNAs in 7 LUAD cell lines and BEAS-2B cells was determined by qRT-PCR. The data are presented as the mean ± standard error of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001. C The relative expression of the candidate DE lncRNA ENST00000609697 in the TCGA dataset. D Kaplan–Meier (KM) survival analysis based on the candidate DE lncRNA ENST00000609697. x axis: overall survival (years); y axis: survival rate. Green and red represent the low and high lncRNA expression groups, respectively
CeRNA regulatory network involving the DE lncRNAs
To further illustrate the potential interactions among the DE lncRNAs, miRNAs DE involved in LUAD, lncRNA–miRNA–mRNA ceRNA networks were constructed with a total of 188 DE lncRNAs, 444 DE miRNAs and 410 DE mRNAs (Additional file 2: Fig. S1E). Moreover, we found that most DE lncRNAs in the ceRNA regulatory network were downregulated. We selected the ceRNA network of the candidate gene ENST00000609697 for further analysis and found that it included 7 miRNAs (hsa-miR-3191-3p, hsa-miR-4731-5p, hsa-miR-598-5p, hsa-miR-6791-5p, hsa-miR-4292, hsa-miR-4446-3p, and hsa-miR-1827) and 20 DE mRNAs (COLGALT2, MYOCD, TNS1, RASL12, CNN1, etc.) (Fig. 6A). The enriched miRNA hsa-miR-4731-5p targeted most DE mRNAs in the ENST00000609697 ceRNA network, indicating that it may play a critical role in LUAD.

Target gene functional annotation of the ENST00000609697 ceRNA regulatory network. A CeRNA network of ENST00000609697. B Bar plot of the top 30 GO classification terms enriched by the target genes, P < 0.05. C BP cnetplot of the target genes, P < 0.001. D BP emapplot of the target genes, P < 0.001. E Relative expression of RASL12 in the TCGA dataset, P < 0.0001. F Survival analysis based on RASL12 in the TCGA dataset, P = 0.034
Functional and survival analyses considering the target DE mRNAs in the ENST00000609697 ceRNA network
We next conducted GO enrichment analysis of the 20 targeted DE mRNAs in three ontologies: BP, CC, and MF. The 30 GO terms most enriched by the 20 targeted DE mRNAs are shown in Fig. 6B. The most enriched GO terms in the BP, CC, and MF categories were smooth muscle cell differentiation, focal adhesion, and actin binding, respectively (Fig. 6B). Most DE mRNAs mapped to the BP category; thus, we generated a BP cnetplot that showed the DE mRNAs associated with the top 10 BP terms; this analysis identified, 4 DE mRNAs (CNN1, FLNC, FOXF1, and MYOCD) (Fig. 6C). FOXF1 and MYOCD are related to multiple biological processes, suggesting that they may be critical genes in LUAD. The BP emapplot showed the overlap** relationship between each pair of terms (Fig. 6D) and suggested that smooth muscle cell differentiation was a very important biological process. To screen ceRNA networks related to LUAD prognosis, we downloaded expression and survival data related to the 20 target DE mRNAs in the ENST00000609697 ceRNA network from the UCSC Xena database. RASL12 expression was downregulated in LUAD (P < 0.0001) (Fig. 6E), and this downregulation was positively correlated with good prognosis (P = 0.034) (Fig. 6F). These results suggested that the ENST00000609697–hsa-miR-6791-5p–RASL12 axis may play a tumor-suppressive role in LUAD.
Discussion
Here, we identified 2819 DE lncRNAs and 2396 DE mRNAs, including 859 upregulated lncRNAs, 1960 downregulated lncRNAs, 757 upregulated mRNAs, and 1639 downregulated mRNAs. More genes were downregulated than upregulated, indicating that downregulated genes may play important roles in the biology of LUAD. To explore the potential mechanisms of the DE genes, we performed GO and KEGG analyses of the aberrantly expressed lncRNAs and mRNAs. GO analysis showed that the DE lncRNAs were enriched mainly in atomic septum development, structural molecule activity conferring elasticity, and embryonic digestive tract morphogenesis and that the DE mRNAs were enriched mainly in condensed chromosome outer kinetochore, cell migration involved in heart development, and regulation of vasculogenesis. However, all the DE lncRNAs DE mRNAs were involved in angiogenesis and cell proliferation. Abnormalities in these two processes are closely related to the occurrence and development of cancers [25, 26]. The KEGG classification results for the DE lncRNAs and mRNAs showed that they were enriched mainly in signal transduction, the immune system, and cancers. Moreover, KEGG pathway enrichment analysis suggested that these DE genes were enriched mainly in vascular smooth muscle contraction, focal adhesion, and the TGF beta signaling pathway, and were also closely related to small cell lung cancer, NSCLC, melanoma, glioma, prostate cancer, thyroid cancer, CRC, and other cancers. According to the previous study, the focal adhesion and the TGF beta signaling pathways play essential roles in cell proliferation, and dysregulation of these two pathways is closely associated with oncogenesis [29, 18, 34]. For example, lncRNA-KRTAP5-AS1 and lncRNA-TUBB2A can competitively bind miR-596 and miR-3620-3p as ceRNAs to promote CLDN4 expression, enhance cell proliferation and invasion, and promote epithelial–mesenchymal transition (EMT) [31]. To identify the potential interactions among the DE lncRNAs, DE miRNAs, and DE mRNAs, we constructed lncRNA–miRNA–mRNA ceRNA networks involving a total of 188 DE lncRNAs, 444 DE miRNAs, and 410 DE mRNAs. Interestingly, most of the DE lncRNAs in the ceRNA regulatory network were downregulated. We then screened the ENST00000609697 ceRNA network, which was downregulated and positively correlated with good prognosis in LUAD. This network contained seven miRNAs (hsa-miR-3191-3p, hsa-miR-4731-5p, hsa-miR-598-5p, hsa-miR-6791-5p, hsa-miR-4292, hsa-miR-4446-3p, and hsa-miR-1827) and 20 DE mRNAs (COLGALT2, MYOCD, TNS1, RASL12, CNN1, etc.). We performed an in-depth analysis of the functions related to the ENST00000609697 ceRNA network and found that the most enriched GO terms in the BP, CC, and MF categories were smooth muscle cell differentiation, focal adhesion, and actin binding, respectively. Smooth muscle cell differentiation is very important for the stability and repair of the vascular system, and abnormalities in this biological process can directly or indirectly affect the growth, proliferation, and migration of tumor cells and the tumor immune microenvironment [35,36,37,38]. Focal adhesions are the center of cellular mechanical sensation and serve as bridges between integrin, the extracellular matrix and the cytoskeleton, which is correlated with the tumor microenvironment. Changes in signal transmission through focal adhesions of malignant cells are very important for tumor cell metastasis [38,39,40]. Actin binding-related proteins participate in the formation of the cytoskeleton and regulate cell adhesion and migration [41]. The proliferation, migration, and invasion of tumor cells are dependent on proteins related to angiogenesis, focal adhesions, and actin binding. Therefore, the ENST00000609697 ceRNA network may play an important role in the tumor microenvironment of LUAD, and its functions are worth further exploration.
Subsequently, we downloaded expression and survival data for the 20 target DE mRNAs in the ENST00000609697 ceRNA network from the UCSC Xena database and found that RASL12 expression was downregulated in LUAD (P < 0.0001) and was positively correlated with good prognosis (P = 0.034). RASL12, a member of the RAS-like GTPase family, is localized in the cytoplasm [42]. However, evidence that RASL12 functions as a small GTP-binding protein is lacking; In fact, studies have reported that RASL12 could be homologous to the RAS-like GTPases RERG, RASL11A, RASL11B, RASL10A and RASL10B, which play tumor-suppressive roles in human cancers [43,44,45]. In addition, a recent study reported that the tumor suppressor RASSF1 can form a complex with RASL12 and recruit RASL12 to microtubules [
where N is the number of genes in the background, M is the number of genes annotated in the background to the biological term, n indicates the length of the gene vector provided in parameter one, and k is the number of genes annotated to the corresponding biological term in set n; and 4) the P value was adjusted for multiple comparisons with the BH (Benjamini and Hochberg) method. The terms identified by this analysis were arranged in descending order according to the enrichment factor value, and the top 30 terms were considered.
Construction of the regulatory network
We selected the top correlated mRNA/lncRNA pairs in the normal and cancer datasets based on the correlation threshold of the 99th percentile of the corresponding overall correlation distribution in both cases. Then, we built up two regression models:
Seed match analysis
A perfect match at positions 2 to 7 of the 5ʹ-end of the mature miRNA sequence (6-mer miRNA seed) is the minimal pairing requirement considered predictive for miRNA target recognition. We used seed match analysis to restrict the above selected lncRNA/miRNA/mRNA triplets to those in which both the lncRNA and mRNA have at least one perfect 6-mer seed match for the shared miRNA.
By integrating the statistical analysis and seed match analysis results, we built the miRNA-mediated interaction (MMI) networks for both normal and cancer tissues. The nodes in these networks represent mRNAs and lncRNAs with highly correlated expression profiles, while the edges represent the miRNAs that mediate the interactions. The regulatory network was visualized using the Cytoscape tool [48] (3.9.0). The configurations (node color, position, and size) were chosen manually.
Statistical analysis
All data are presented as the mean ± standard error. Paired samples compared using a paired two-tailed Student’s t test. Multiple group comparisons were performed with an one-way ANOVA followed by Dunnett’s multiple comparison test. Statistical analyses were performed with GraphPad Prism 5 (GraphPad Software, USA). P values less than 0.05 were considered to indicate statistical significance.
