
Abstract
Background
An orphan disease is any disease that affects a small percentage of the population. Orphan diseases are a great burden to patients and society, and most of them are genetic in origin. Unfortunately, our current understanding of the genes responsible for inherited orphan diseases is still quite limited. Develo** effective computational algorithms to discover disease-causing genes would help unveil disease mechanisms and may enable better diagnosis and treatment.
Results
We have developed a novel method, named as DIGNiFI (Disease causIng GeNe FInder), which uses Protein-Protein Interaction (PPI) network-based features to discover and rank candidate disease-causing genes. Specifically, our approach computes topologically similar genes by taking into account both local and global connected paths in PPI networks via Direct Neighbors and Local Random Walks, respectively. Furthermore, since genes with similar phenotypes tend to be functionally related, we have integrated PPI data with gene ontology (GO) annotations and protein complex data to further improve the performance of this approach. Results of 128 orphan diseases with 1184 known disease genes collected from the Orphanet show that our proposed methods outperform existing state-of-the-art methods for discovering candidate disease-causing genes. We also show that further performance improvement can be achieved when enriching the human-curated PPI network data with text-mined interactions from the biomedical literature. Finally, we demonstrate the utility of our approach by applying our method to identifying novel candidate genes for a set of four inherited retinal dystrophies. In this study, we found the top predictions for these retinal dystrophies consistent with literature reports and online databases of other retinal dystrophies.
Conclusions
Our method successfully prioritizes orphan-disease-causative genes. This method has great potential to benefit the field of orphan disease research, where resources are scarce and greatly needed.
Similar content being viewed by others


Background
The US Rare Disease Act of 2002 defined a rare disease, also referred to as an orphan disease, as any disease that affects fewer than 200,000 inhabitants, equivalent to approximately 6.5 patients out of 10,000 inhabitants [1]. There are an estimated 8000 orphan diseases, and most of them are genetic in origin. Orphan diseases are a great burden to patients and society because they commonly afflict people early in life and persist throughout the lifetime. Some are even life-threatening [2, 3]. Discovering genes causing these diseases would unveil disease mechanisms and may enable better diagnosis and treatment. Unfortunately, our current understanding of the genes responsible for genetic orphan diseases is still quite limited [4]. In addition, even though the advent of next-generation sequencing has yielded great advances in our ability to collect data about patients with rare diseases, successfully sorting through this information to correctly identify the causal genes remains challenging [5]. Therefore, develo** effective computational algorithms for the prioritization of candidate genes is a critical step in the research pipeline. Several earlier studies have shown that genes related to similar disease phenotypes tend to be functionally related, since genes execute their functions by interacting with one another through such means as sharing similar expression profiles, participating in signal tranduction mechanisms [6–8]. Moreover, researches have also shown that genes associated with phenotypically close disorders are likely to directly or indirectly interact with each other in the protein-protein interaction (PPI) network [8–11]. Based on this core concept and the principle of “guilt-by-association” [12], we propose a novel method to explore PPI networks and discover disease-causing genes.
Many computational approaches have been developed and applied to prioritize candidate disease genes from PPI networks [13–20]. Network-based disease-gene prioritization approaches can be broadly grouped into two categories: local similarity measures [14, 19] and global similarity measures [13, 15]. Local similarity measures consider whether two genes are directly connected or have a shorter path in the PPI network. However, many disease-causing genes don’t have such local connections and can only be connected through distant paths in PPI networks. For this reason, some groups have used global similarity measures obtained via various methods such as Random Walk with Restarts (RWR) to calculate the similarity between candidate genes and known disease genes [13]. Although these kinds of methods accurately capture global topological features of PPI networks, most of them require extensive computation. For these reasons, accurately identifying candidate genes through PPI networks remains challenging. In this paper, we propose a method, DIGNiFI, that calculates the topological similarity between two genes by considering local features based on shared Direct Neighbors and global features by Local Random Walks (LRW) [21].
Meanwhile, because human-curated PPI data contain various false-positive and false-negative interactions [22–24], integrating multiple resources such as gene expression profiles or GO annotations, is an alternative way to reduce the potential bias of using PPI data as a single resource for disease gene prioritization [18, 20, 25]. In addition, there is increasing evidence in genetic and molecular biology that protein complexes and pathways affect the interactions within groups of genes, perturbations of which lead to similar diseases [26]. In this work, we integrate GO annotations and protein complex data (DIGNiFI+SimBio) to further improve our proposed algorithm DIGNiFI. Also we apply these methods on an enriched network [27] that we generated by combining gene relations obtained based on proteins’ co-occurrences in biomedical literature and PPI interactions from biological experiments. All the algorithms were tested on 128 orphan diseases with at least five known genes downloaded from Orphanet [28]. The results demonstrate that our approach outperforms four state-of-the-art algorithms: VS [14], RWR [13], SPranker and SPGOranker [18]. VS method uses shortest paths to assess the closeness between two genes and RWR uses Random Walk with Restart to measure the distance between two genes, while SPranker uses shortest paths with weights to calculate the similarity between two genes and SPGOranker combines SPranker with GO functional annotations.
Furthermore, we apply our method to predict potential causative genes for several orphan eye diseases. Our top predictions include many genes with known associations with similar eye diseases and are consistent with literature reports and online databases. This case investigation of our method demonstrates its capability to discover causative genes for orphan diseases and suggests that other prioritized genes from our approach may be excellent candidates for further investigation.
Methods
DIGNiFI algorithm
The core assumption of disease gene prioritization from a PPI network is that genes that share topological similarities tend to be associated with phenotypically close disorders and may cause the same or similar diseases [8, 11, 29]. Such a “guilt by association” principle has been widely used to prioritize candidate disease genes. Hence, the most important task in using PPI network is measuring the similarity between known genes and candidate genes. In order to rank the candidate genes, we use two different ways to calculate the similarity: one is designed for directly connected genes and the other is for indirectly connected genes in the PPI network.
A PPI network can be presented as a graph G(V,E,W), where a set of nodes (V) denotes proteins together while a set of edges (E) denotes interactions between proteins with different edge weights (W). Given a protein v∈V, Γ v represents the combination of v’ neighbors and v. From a topological view, if two genes share more common direct neighbors, those two genes are likely to be more similar. Hence, given a protein pair v i and v j , we calculate the similarity between them by using Eq. 1.
The value of DN(v i ,v j ) is defined as:
where w(v i ,v j )=1 if v i and v j directly connect with each other or if v i =v j , otherwise w(v i ,v j )=0. \(K_{v_{i}}\) denotes the total edge weights that link to v i , and we use the maximum of K in order to depress the hub node effect. The value of \(LRW_{v_{i}v_{j}}\) is derived by Eq. 3 according to [21].
where M is the number of links in the network and \({\pi }_{v_{i}v_{j}}(t)\) is the v j -th value of \(\boldsymbol {\pi }_{v_{i}}(t)\) and \(\boldsymbol {\pi }_{v_{i}}(t)\) is calculated by
in which, π x (0) is a N∗1 vector (N is the number of nodes in the network) which the v i -th is equal to 1 and others are 0. P is the transition probability matrix, with \({P}_{v_{i}v_{j}}=a_{v_{i}v_{j}}/k_{v_{i}}\) representing the probability that a random walker staying at node v i will walk to v j in the next step, where \(a_{v_{i}v_{j}}\) equals 1 if v i and v j are connected, 0 otherwise. For two connected nodes, we use DN, which emphasizes the similarity of common direct neighbors, to calculate the similarity between them. In addition, we also consider that this approach may result in hub nodes receiving inappropriately high ranks, since they are connected to more nodes but are not necessarily the most directly similar genes. So we use maximum weight to penalize the hub nodes. At the same time, we use LRW to calculate the similarity between two indirectly connected nodes. One difficulty with general random-walk-based similarity measures is that they sensitively depend on parts of the network far away from the source nodes [30]. For example, the walker has a certain probability to go too far away from a source node to a target node even though they may in reality be close to each other. Using the LRW method can counteract this dependence and assign high similarity scores to the target node and the nodes nearby. Besides, the t step Local Random Walk algorithm has lower computational complexity than other random walk based algorithms and is suitable for scale and sparse networks [21]. As most disease genes connect with each other through calculable steps and as the PPI network is a large-scale yet sparse network [31, 32], LRW is a high-performance way to calculate the similarity between genes in the PPI network.
For a given disease d, if \(S_{d_{k}}\) denotes the set of known genes, then the probability of a new candidate gene v c to be a causal gene is evaluated by the sum similarity scores between all known genes and the candidate gene, as shown in Eq. 5:
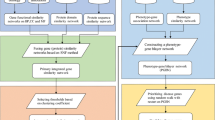
After calculating the total score, we rank candidate genes of the given disease by their total scores. Figure 1 shows the flow chart of using DIGNiFI to prioritize disease causing genes for a query disease. As the similarity scores of DN and LRW can be pre-calculated, the complexity of ranking candidates genes depends only on the number of known genes when given a new disease.

The flow chart of using DIGNiFI to prioritize diseases causing genes
Integration with biological resources
It is well known that PPI data contain various false positive and false negative links. Therefore, integrating different data resources with PPI data should reduce the bias of using PPI data as a single resource and increase the ability of the PPI network to prioritize disease-causing genes. Recent research has demonstrated that genes with similar phenotypes often share common molecular signatures such as biological function, as measured by GO annotations [8]. Also, protein complex data is distinct from PPI network data, with clear, biologically relevant distinctions. For example, PEX26, PEX16 and PEX3 are three causal genes of Zellweger Syndrome. These genes don’t have any direct interaction in the PPI network, but do form a real protein complex. Hence, we integrate GO annotations and protein complex data to further improve our method.
Gene ontology annotation
The Gene Ontology project [33] provides a collection of well-defined biological terms for annotating genes and describing the characteristics of their gene products. GO annotation terms cover three separate fields: biological process, molecular function, and cellular components [34]. Many computational methods have used semantic similarity to calculate the similarity between two concepts in a taxonomy [35]. We employed a modification of a previous method [36] to calculate two genes’ semantic similarity by considering the number of common GO terms and how many genes the common GO terms have annotated. Specifically, we calculate two genes’ similarity based on their shared GO terms including biological process, molecular function and cellular component GO terms. For a given GO term, we define the annotation size of a GO term as the number of genes with that GO term. We then calculate the semantic similarity between two genes by the annotation size of their common GO terms. Thus, if two genes share a smaller annotation size of GO term, they are considered functionally more similar.
To describe the algorithm clearly, we first give some definitions. For a given gene v i , suppose it is annotated with m different GO terms. S k (v i ) denotes a set of annotated genes with the GO term g k , whose annotation set includes v i , where 1≤k≤m. Suppose n is the number of common GO terms between gene v i and v j , where n≤m. S k (v i ,v j ) denotes a set of annotated genes on GO term g k whose annotation set includes both v i and v j , where k≤n. Then, the semantic similarity of two genes based on GO annotations is calculated by the following formula:
where min k |S k (v i ,v j )| is the minimum size of S k (v i ,v j ) and S max is the maximum size of annotation among all GO terms.
Protein complex
Protein complexes are direct manifestations of the biologic interconnectivity of genes. It is likely that variants of genes whose protein products form complexes together may lead to similar disease phenotypes. Indeed, protein complexes have already been successfully used to predict disease-causing genes [37, 38]. However, these approaches overlook the information of the actual protein complexes by only using formed protein complexes based on topological properties (neighbors or densely connected subsets). Furthermore, these previous studies did not consider any of the unique characteristics of each protein complex. Many groups have demonstrated that dense subgraphs in a PPI network generally correspond to protein complexes [39, 40], and some studies show that if the nodes of a subgraph have more internal weight (or edges) than external weight (or edges), it will be more likely to form a group [41]. Thus the density and internal weight ratio of protein complex in a PPI network can be an index for the richness of protein interactions within the complex. In other words, proteins are more similar if they are in a more dense protein complex. Considering the two issues, we use the internal weight ratio [42] and the density to assign a network reliability score to an actual complex C k . The formula is shown by Eq. 7:
where, w in(C k ) is the total edges’ weight within a complex and w bound(C k ) is the total weight of edges that connect the complex with the rest of the network. The density of a protein complex C k is defined as Eq. 8:
where \(E_{C_{k}}\) and \(V_{C_{k}}\) denote the edges and nodes in the complex respectively. Then, the Score(C k ) can quantify the richness and reliability of the interactions with C k .
If two genes are in M same protein complexes, the similarity score between them is calculated as:
Finally, in order to integrate biological similarity (SimBio) with topological similarity (DIGNiFI), parameters α and β are used. The total score of a candidate gene with a known gene is calculated as:
Then, a candidate gene of a query disease is ranked by summing up the similarity scores between the candidate gene and the known genes of that disease.
Results and discussion
Data sources
Protein-protein interaction (PPI) data were downloaded from release 9 of the Human Protein Reference Database (HPRD) [43]. After removing duplicates and self-linked interactions, we obtained 9453 human genes and 36,867 interactions. Orphan diseases with causal genes were downloaded from Orphanet [28]. We selected all orphan diseases that had at least five causal genes found in the protein interaction network, resulting in 128 diseases and 1184 total genes.
The PPI network contains a number of false negative interactions because many interactions remain undetected by biological experiments. Nevertheless, the biomedical literature contains descriptions of many PPIs that are not catalogued in HPRD. For this reason in this paper, we also used the gene2pubmed dataset [44], which contains curated information about gene descriptions in PubMed articles, to test if combining protein relations mined from literature would be helpful for prioritization of disease-causing genes. We assumed that if mentions of different genes co-occur in an abstract, those genes are likely to have some types of interactions (direct or indirect). Note that we chose simple co-occurrence instead of advanced text mining techniques because relation extraction between bio-concepts remains challenging [45–47]. Thus to ensure high quality results from literature, we set a rather high threshold (the same pair needs to occur in 30 or more PubMed articles in this study), resulting in the selection of 16,118 gene interactions, 9600 of which overlapped with interactions in HPRD and 6518 did not. We merged these 6518 literature-mined interactions with the existing HPRD data.
Experimental setting and evaluation criteria
Leave-one-out cross-validation was used to compare the proposed method with four state-of-the-art methods: RWR (with restart probability set to 0.8), VS, SPranker and SPGOranker (combining SPranker with GO functional annotations). Furthermore, we also compared DIGNiFI with LRW which calculates the similarity between two genes using Local Random Walk no matter whether these two genes are directly connected or not. In each round of cross-validation, one causal gene within an orphan disease, as the target gene, was removed. The remaining known causative genes for that disease were used as seed nodes, and each method was evaluated by the number of overall successes of ranking the target gene among top k. Specially, if the similarity score is equal to zero, then the rank of that candidate gene will be the size of the total candidate gene set. Considering the fact that the predicted top-ranked results are more important in practice, we utilized k values ranging from 1 to 10 in this paper, and the ratio of successful validation trails was used as the criterium for determining the “success rate”. In each cross validation trial, one target gene mixed with 99 randomly selected genes formed a set of 100 candidate genes. The step t of DIGNiFI is 3 as suggested by [48], and the default values of α and β in this paper are both to be 0.1.
Experimental results
The top-k-ranking results of each algorithm on the HPRD dataset are presented in Fig. 2. To perform these experiments, we manipulated the values of α and β to evaluate the contribution of each of these DIGNiFI modifications separately. Kee** β fixed as 0.0, only GO information is used (DIGNiFI+SimGO) and the best performance is obtained when α=0.1. While when α=0.0 only complex information is used (DIGNiFI+SimCOM) and the best performance is gained by setting β to 0.3. When k=1, the DIGNiFI+SimBio achieved the best performance with α=0.1 and β=0.1, its success rate is 48.65% (576/1184). Out of 1184 genes, DIGNiFI+SimGO successfully prioritized 553 (46.71%), and LRW+SimCOM successfully prioritized 414 (34.97%). By contrast, the success rates of DIGNiFI, LRW, VS, RRW and SPranker were 34.54% (409/1184), 30.74% (364/1184), 31.67% (375/1184), 29.48% (349/1184) and 31.84% (377/1184), respectively. Among all these five topological-feature-based methods, DIGNiFI performs best. The SPGOranker prioritization method, which also incorporates GO functional information, correctly predicted 421 genes, achieving a success rate of 35.56% and outperforming all others except DIGNiFI+SimGO and DIGNiFI+SimBio, which still surpassed SPGOranker by 34.36% and 36.81% respectively. Figure 2 shows that the DIGNiFI+SimBio consistently performs best throughout a range of k values from 1 to 10. Interestingly, these experiments also demonstrate that although VS performs better than RWR when k equals to 1, its performance only marginally improves with increasing k value. This is because VS only calculates two-node similarities by taking into account only proteins with direct interactions or shared neighboring nodes. If causal genes have more than one hop or step between them, VS is not able to identify their similarity. Furthermore, although ND-LRW+SimCOM does not correctly identify as many genes as DIGNiFI+SimGO and SPGOranker which both use GO information, it still outperforms the other methods that only use topological information, especially when k=1. These results demonstrate the effectiveness of the DIGNiFI algorithms and integration of biological resources improves the ability to detect disease-causing genes.

Performance comparison of different algorithms
Analysis of the results when k=1
To further determine the performance of these methods, we analyzed the intersection of each result with k=1. The intersection results are shown in Table 1. Both the columns and rows represent the method used. The values in the table reflect the number of overlap** predictions generated by the corresponding column and row methods. DIGNiFI+SimBio understandably covers a high percentage of other methods since it identifies the greatest number of true disease-causing genes, including more than 100 different genes that were not detected by any other method. DIGNiFI+SimCOM with only 414 successful top-one predictions, surprisingly covers nearly 85% of the genes identified by other topological-feature-based methods. This indicates that both protein complex and GO annotations can help improve the performance for prioritization of disease-causing genes from another side. Interestingly, this table also demonstrates that different methods succeeded in identifying different genes. In order to determine if the results are significantly different from each other, we use two-tailed Student t test to compare the ranking results. The p-values between the ranking results from DIGNiFI+Simbio and other four methods are less than 0.05 (SPGOranker: 9.65E−26, SPranker: 1.51E−27, RWR: 3.58E−23, VS: 1.39E−70), which suggests that the differences between our methods and other state-of-the-art methods are not by chance.
Analysis of the effect of α and β
To analyze the effect of parameters α and β, we tested our prediction algorithms using values of α and β from 0.0 to 1.0 with 0.1 increments. The results with k=1 are shown in Table 2. When the parameters both equal to 0.0, the result is derived by DIGNiFI only. When α=0.0, the results are obtained by DIGNiFI+SimCOM while when β=0.0 the results are from DIGNiFI+SimGO. From the table, we can see that the results obtained from integrating PPI networks with biological information are much better than the results obtained from using topological similarity alone. Furthermore, the combined approach using both GO annotations and topological similarity outperforms the GO annotations approach. This indicates that functional and topological similarity contribute unique information to gene prioritization. Table 2 demonstrates that the success ratio for top one prediction is very low when using protein complex data alone. This may be due to the relatively small number of known protein complexes. When fusing protein complex data and topological similarity, although the results increase only slightly and some of them are not as good as using DIGNiFI, the best result of DIGNiFI+SimCOM show that it still can detect 82 different true disease causal genes than DIGNiFI+SimBio according to Table 1. These results show that even though protein complex data is not as rich as PPI network and GO annotation data, integrating real protein complex data still helps to improve prioritization of disease causal genes. This indicates that we may obtain improvements in the ability of prioritizing disease-causing genes with enhancement of protein complex data.
The results on literature enriched PPI network
For the reasons described previously in “Data sources” subsection, we used data from gene2pubmed to enrich the PPI network from HPRD. Figure 3 shows the best results of DIGNiFI and DIGNiFI+SimBio on both the original and enriched networks. The DIGNiFI and DIGNiFI+SimBio prediction algorithms perform consistently better on the enriched network than on the original network. These results indicate that the quality of PPI network affects the performance of gene prioritization. Since the performances of these methods improve after integrating even simple co-occurrence-based literature protein interactions into the PPI network, it is likely that further, systematic enhancements to the existing PPI network will result in continued improvements in disease gene prioritization performance.

Performance comparison of different networks
Case study: prioritizing causal genes for inherited eye diseases
We further examined the capabilities of our method DIGNiFI+SimBio by predicting genes for a collection of four eye diseases, all of which involve degeneration of the retina: Retinitis pigmentosa (RP), Leber congenital amaurosis(LCA), Usher syndrome Type 1, and Congenital stationary night blindness (CSNB). All these diseases belong to a collection of rare, inheritable disorders called retinal dystophies. We include a brief description of each in Table 3. Orphanet contains lists of known genetic associations with these disorders, but another manually curated resource, the Retinal Information Network (RetNet), contains more comprehensive information about the genes related to these and other retinal dystrophies. We used the known genes listed in Orphanet as seed genes to predict additional genes that might be associated with these diseases. Specifically, for each disease, we ranked all genes in PPI network except the known genes in order to obtain the top ten predictions. We then compared our predictions with genes in RetNet to evaluate their validity.
As mentioned previously, a key assumption of PPI-network-based approaches to gene prioritization is that genes that share topological similarities tend to be associated with phenotypically close disorders. Therefore, we hypothesized that the genes that DIGNiFI+SimBio would predict from the Orphanet seed genes for a given retinal dystrophy would overlap with either the known genes listed in RetNet for that condition or the known genes for other retinal dystrophies. There were three possible outcomes of interest for each disorder we investigated. DIGNiFI+SimBio-predicted genes might (1) overlap with gene curations in RetNet for that disorder; (2) overlap with genes known to cause similar retinal disorders; or (3) not overlap at all with any known associations between genes and retinal dystrophes. It was also possible that DIGNiFI+SimBio might not prioritize genes that nevertheless had known associations in RetNet. Table 3 contains the results of this analysis.
The genes in the third column of Table 3 are hypothetical genes listed in order of their likelihood to be associated with a given disease as determined by DIGNiFI+SimBio. Two of the gene predictions generated by DIGNiFI+SimBio overlapped with known genes in RetNet. These genes, DFNB31 and USH2A, were the top two predictions for Usher Syndrome, Type 1. It is interesting to note that both genes are actually known to be associated with Usher Syndrome, Type 2. Although these specific hypotheses would thus not likely have practical utility for researchers or clinicians, they offer striking support for the validity of DIGNiFI+SimBio’s predictions. Furthermore, nearly all predicted genes for each retinal dystrophy overlapped with RetNet lists of genes for other retinal dystrophies. This overlap demonstrates that it is possible to use the topological features of PPI networks to identify functionally related and likely interdependent genes. The fourth column of Table 3 contains six genes that are listed in RetNet but not in Orphanet as having an association with the four retinal dystrophies we investigated. These genes were not in the top 10 predictions from DIGNiFI+SimBio but were in the PPI network. In investigating these genes, we found their ranks were 17(ARL3), 13 (HK1), 20(CEP250), 11(HARS), 33(GNB3) and 14(RDH5) respectively.
In addition to comparing our predictions with curations in RetNet, we also conducted a literature review for each disorder and its set of predicted genes. We performed this review by executing semantic searches for the relevant genes and diseases using entity-tagging tools in PubTator [49, 50]. Our search identified specific support for 10 of the 40 DIGNiFI + SimBio gene prioritizations, including at least one for each of the retinal dystrophies we studied. In some cases, the support was very strong. For example, PMID: 26900326 is a recent paper that reports an association between the gene MKKS, which has traditionally been associated with Bardet-Biedl syndrome, and retinitis pigmentosa. DIGNiFI + SimBio prioritized MKKS among the top ten novel genes from PPI most likely to contribute to retinitis pigmentosa. Although retinitis pigmentosa is one of the findings of Bardet-Biedl syndrome, the case reported in this study is unique because the retina findings resulting from this mutation caused retinitis pigmentosa in isolation of the other findings of Bardet-Biedle syndrome. Table 4 contains the results of the most relevant support that we identified in our literature investigation of the predicted genes.
Although we identified support for many gene predictions through the literature and curations in RetNet, many genes remained without representation in any of these knowledge sources. These truly novel genetic hypotheses are strong leads for future research and discovery.
Conclusion
In this paper, we propose a new algorithm, DIGNiFI, to prioritize causal genes for inherited orphan diseases. DIGNiFI considers both local and global features of genes in the PPI network and specifically uses Local Random Walks to identify global features. Leave-one-out cross-validation experiments with DIGNiFI show that DIGNiFI outperforms other algorithms that use topological features especially with ranking the top gene. We also explored the benefits of incorporating biological information from GO annotations and protein complex data into PPI network predictions. The resulting algorithm, DIGNiFI+SimBio, does indeed attain enhanced performance in predicting disease-causing genes. Furthermore, we reconstructed a PPI network by merging protein interactions from HPRD with protein interactions extracted from the literature using co-occurrence. Test results using DIGNiFI and DIGNiFI+SimBio on this text-mining enriched PPI network indicate not only that the sparseness of the PPI network limits gene prioritization but also that PPI relationships mined from biomedical literature can improve the quality of the PPI network and enhance gene-prioritization performance. Lastly, we use DIGNiFI+SimBio to predict genes involved in a set of four inherited retinal dystrophies. We found near-universal involvement of the predicted genes with retinal diseases and identified supporting literature for several of the hypothesized gene-disease associations. Taken together, these results demonstrate the relevance of our prediction method and indicate its potential utility in the field of orphan disease research, where resources are scarce and greatly needed.
References
Dear JW, Lilitkarntakul P, Webb DJ. Are rare diseases still orphans or happily adopted? The challenges of develo** and using orphan medicinal products. Br J Clin Pharmacol. 2006; 62(3):264–71.
Stolk P, Willemen MJ, Leufkens HG. Rare essentials: drugs for rare diseases as essential medicines. Bull World Health Organ. 2006; 84(9):745–51.
Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008; 371(9629):2039–41.
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013; 14(10):681–91.
Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011; 144(6):986–98.
Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002; 3(10):779–89.
Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc Natl Acad Sci. 2008; 105(11):4323–8.
Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011; 12(1):56–68.
Oti M, Brunner HG. The modular nature of genetic diseases. Clin Genet. 2007; 71(1):1–11.
Wysocki K, Ritter L. Diseasome An Approach to Understanding Gene–Disease Interactions. Annu Rev Nurs Res. 2011; 29(1):55–72.
Tang H, Zhong F, **e H. A quick guide to biomolecular network studies: construction, analysis, applications, and resources. Biochem Biophys Res Commun. 2012; 424(1):7–11.
Li J, Zheng S, Chen B, Butte AJ, Swamidass SJ, Lu Z. A survey of current trends in computational drug repositioning. Brief Bioinform. 2016; 17(1):2–12.
Köhler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008; 82(4):949–58.
Zhu C, Kushwaha A, Berman K, Jegga AG. A vertex similarity-based framework to discover and rank orphan disease-related genes. BMC Syst Biol. 2012; 6(Suppl 3):S8.
Le DH, Kwon YK. Neighbor-favoring weight reinforcement to improve random walk-based disease gene prioritization. Comput Biol Chem. 2013; 44:1–8.
Jiang L, Edwards SM, Thomsen B, Workman CT, Guldbrandtsen B, Sørensen P. A random set scoring model for prioritization of disease candidate genes using protein complexes and data-mining of GeneRIF, OMIM and PubMed records. BMC Bioinforma. 2014; 15(1):315.
Valentini G, Paccanaro A, Caniza H, Romero AE, Re M. An extensive analysis of disease-gene associations using network integration and fast kernel-based gene prioritization methods. Artif Intell Med. 2014; 61(2):63–78.
Li M, Li Q, Ganegoda GU, Wang J, Wu F, Pan Y. Prioritization of orphan disease-causing genes using topological feature and GO similarity between proteins in interaction networks. Sci China Life Sci. 2014; 57(11):1064–71.
Hsu CL, Huang YH, Hsu CT, Yang UC. Prioritizing disease candidate genes by a gene interconnectedness-based approach. BMC genomics. 2011; 12(3):1.
Liu B, ** M, Zeng P. Prioritization of candidate disease genes by combining topological similarity and semantic similarity. J Biomed Inform. 2015; 57:1–5.
Liu W, Lü L. Link prediction based on local random walk. EPL (Europhysics Letters). 2010; 89(5):58007.
Von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, et al.Comparative assessment of large-scale data sets of protein–protein interactions. Nature. 2002; 417(6887):399–403.
Sprinzak E, Sattath S, Margalit H. How reliable are experimental protein–protein interaction data. J Mol Biol. 2003; 327(5):919–23.
Wodak SJ, Vlasblom J, Turinsky AL, Pu S. Protein–protein interaction networks: the puzzling riches. Curr Opin Struct Biol. 2013; 23(6):941–53.
Zhang SW, Shao DD, Zhang SY, Wang YB. Prioritization of candidate disease genes by enlarging the seed set and fusing information of the network topology and gene expression. Mol BioSyst. 2014; 10(6):1400–8.
Suthram S, Dudley JT, Chiang AP, Chen R, Hastie TJ, Butte AJ. Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput Biol. 2010; 6(2):e1000662.
Lu Z, Hirschman L. Biocuration workflows and text mining: overview of the BioCreative 2012 Workshop Track II. Database. 2012; 2012:bas043.
Aymé S. Orphanet, an information site on rare diseases. Soins; la revue de ré, férence infirmière. 2003; 672:46.
Wolfe CJ, Kohane IS, Butte AJ. Systematic survey reveals general applicability of. BMC Bioinforma. 2005; 6(1):227.
Liben-Nowell D, Kleinberg J.The link-prediction problem for social networks. J Am Soc Inf Sci Technol. 2007; 58(7):1019–31.
Tan K, Ideker T. Protein interaction networks. Biol Netw. 2007; 133:133.
Hao T, Peng W, Wang Q, Wang B, Sun J. Reconstruction and Application of Protein–Protein Interaction Network. Int J Mol Sci. 2016; 17(6):907.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al.Gene Ontology: tool for the unification of biology. Nat Genet. 2000; 25(1):25–29.
Consortium GO, et al.Gene ontology consortium: going forward. Nucleic Acids Res. 2015; 43(D1):D1049—D56.
Resnik P. Using information content to evaluate semantic similarity in a taxonomy. In: Proceedings of the 14th International Joint Conference on Artificial Intelligence: 1995. p. 448–53.
Xu B, Lin H, Yang Z.Ontology integration to identify protein complex in protein interaction networks. Proteome Sci. 2011; 9(1):1.
Lage K, Karlberg EO, Størling ZM, Olason PI, Pedersen AG, Rigina O, et al.A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol. 2007; 25(3):309–16.
Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010; 6(1):e1000641.
Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004; 5(2):101–13.
Luo F, Yang Y, Chen CF, Chang R, Zhou J, Scheuermann RH. Modular organization of protein interaction networks. Bioinformatics. 2007; 23(2):207–14.
Radicchi F, Castellano C, Cecconi F, Loreto V, Parisi D. Defining and identifying communities in networks. Proc Natl Acad Sci U S A. 2004; 101(9):2658–63.
Nepusz T, Yu H, Paccanaro A. Detecting overlap** protein complexes in protein-protein interaction networks. Nat Methods. 2012; 9(5):471–2.
Prasad TK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, et al.Human protein reference database–2009 update. Nucleic Acids Res. 2009; 37(suppl 1):D767—D72.
NCBI RC. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016; 44(D1):D7.
Baumgartner Jr WA, Lu Z, Johnson HL, Caporaso JG, Paquette J, Lindemann A, et al.An integrated approach to concept recognition in biomedical text. In: Proceedings of the Second BioCreative Challenge Evaluation Workshop. Madrid: CNIO: 2007. p. 257–71.
Arighi CN, Lu Z, Krallinger M, Cohen KB, Wilbur WJ, Valencia A, et al.Overview of the BioCreative III workshop. BMC Bioinforma. 2011; 12(8):1.
Wei CH, Peng Y, Leaman R, Davis AP, Mattingly CJ, Li J, et al.Assessing the state of the art in biomedical relation extraction: overview of the BioCreative V chemical-disease relation (CDR) task. Database. 2016; 2016:baw032.
Liu W, Lü L. Link prediction based on local random walk. EPL (Europhysics Letters). 2010; 89(5):58007.
Wei CH, Kao HY, Lu Z. PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Res. 2013:gkt441.
Wei CH, Harris BR, Li D, Berardini TZ, Huala E, Kao HY, et al.Accelerating literature curation with text-mining tools: a case study of using PubTator to curate genes in PubMed abstracts. Database. 2012; 2012:bas041.
Funding
This research was supported by the NIH Intramural Research Program, National Library of Medicine (ZL) and the NIH Graduate Partnerships Program (XL was a visiting student at NLM) and the grants from the Natural Science Foundation of China (No. 61070098, 61272373, 61340020, 61572102 and 61572098), Trans-Century Training Program Foundation for the Talents by the Ministry of Education of China (NCET-13-0084) and the Fundamental Research Funds for the Central Universities (No. DUT13JB09 and DUT14YQ213) and the Major State Research Development Program of China (No. 2016YFC0901902). This research was also supported by the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, the Howard Hughes Medical Institute, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors. No funds from the Doris Duke Charitable Foundation were used to support research that used animals. Publication charges were funded by the NIH Intramural Research Program, National Library of Medicine (ZL) and the NIH Graduate Partnerships Program.
Availability of Data and Materials
ftp://ftp.ncbi.nlm.nih.gov/pub/lu/DIGNiFI/.
Authors’ contributions
XL and ZL conceived the project, and XL carried out all experiments and drafted the manuscript. MS participated in analyzing experimental results. ZL, ZY, HL and MS revised this manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
About this supplement
This article has been published as part of BMC Systems Biology Volume 11 Supplement 3, 2017: Selected original research articles from the Third International Workshop on Computational Network Biology: Modeling, Analysis, and Control (CNB-MAC 2016): systems biology. The full contents of the supplement are available online at http://bmcsystbiol.biomedcentral.com/articles/supplements/volume-11-supplement-3.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, X., Yang, Z., Lin, H. et al. DIGNiFI: Discovering causative genes for orphan diseases using protein-protein interaction networks. BMC Syst Biol 11 (Suppl 3), 23 (2017). https://doi.org/10.1186/s12918-017-0402-8
Published:
DOI: https://doi.org/10.1186/s12918-017-0402-8