
Abstract
Background
CRISPR-Cas12a (formerly Cpf1) is an RNA-guided endonuclease with distinct features that have expanded genome editing capabilities. Cas12a-mediated genome editing is temperature sensitive in plants, but a lack of a comprehensive understanding on Cas12a temperature sensitivity in plant cells has hampered effective application of Cas12a nucleases in plant genome editing.
Results
We compared AsCas12a, FnCas12a, and LbCas12a for their editing efficiencies and non-homologous end joining (NHEJ) repair profiles at four different temperatures in rice. We found that AsCas12a is more sensitive to temperature and that it requires a temperature of over 28 °C for high activity. Each Cas12a nuclease exhibited distinct indel mutation profiles which were not affected by temperatures. For the first time, we successfully applied AsCas12a for generating rice mutants with high frequencies up to 93% among T0 lines. We next pursued editing in the dicot model plant Arabidopsis, for which Cas12a-based genome editing has not been previously demonstrated. While LbCas12a barely showed any editing activity at 22 °C, its editing activity was rescued by growing the transgenic plants at 29 °C. With an early high-temperature treatment regime, we successfully achieved germline editing at the two target genes, GL2 and TT4, in Arabidopsis transgenic lines. We then used high-temperature treatment to improve Cas12a-mediated genome editing in maize. By growing LbCas12a T0 maize lines at 28 °C, we obtained Cas12a-edited mutants at frequencies up to 100% in the T1 generation. Finally, we demonstrated DNA binding of Cas12a was not abolished at lower temperatures by using a dCas12a-SRDX-based transcriptional repression system in Arabidopsis.
Conclusion
Our study demonstrates the use of high-temperature regimes to achieve high editing efficiencies with Cas12a systems in rice, Arabidopsis, and maize and sheds light on the mechanism of temperature sensitivity for Cas12a in plants.
Similar content being viewed by others

Background
Many genome editing outcomes are achieved through sequence-specific nucleases (SSNs) which generate targeted DNA double-stranded breaks (DSBs). SSNs such as zinc-finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) were widely applied in genome editing in plants [1]. However, in recent years, these SSNs have been overtaken by CRISPR (clustered regularly interspaced short palindromic repeats) systems with Cas9 or Cas12a (formerly Cpf1) nucleases that mediate DNA targeting through guide RNAs, which are easy to engineer [2,3,4,5,6,7,8].
CRISPR-Cas12a is an RNA-guided endonuclease that has provided new opportunities in genome editing through its distinct features from the commonly used CRISPR-Cas9 system. First, the PAM requirement for Cas12a is “TTTV” or “TTV,” which is advantageous for targeting promoters and other AT-rich sites in gene coding regions [3]. Cas12a requires only a ~ 43-nt guide CRISPR RNA (crRNA) and processes pre-crRNAs into mature RNAs, an ability that endows Cas12a systems with a natural multiplexing ability [9,10,11,12]. Additionally, Cas12a creates 4–5 bp staggered overhangs which may potentially facilitate gene replacement. Finally, Cas12a, unlike other nucleases such as Cas9, is not toxic in some organisms such as Chladymonas [1: Figure S6) and Sanger sequencing (Fig. 3). The editing frequencies in T0 lines at OsDEP1 and OsROC5 were 77.8% and 92.8%, respectively (Fig. 3a and Additional file 1: Figure S6). For OsDEP1, 61.9% mutants were biallelic, and for OsROC5, 46.2% mutants were biallelic. Nearly 100% mutations in these T0 lines were deletions (Fig. 3b), which was consistent with the deletion size analysis in protoplasts (Fig. 2 and Additional file 1: Figure S4). The data suggest that when a high-temperature regime is applied during rice tissue culture, AsCas12a can reliably generate targeted mutations at high frequencies.

High-temperature treatment resulted in high-frequency genome mutations by AsCas12a in stably transformed rice. a Table of mutation rates and observed genotypes at OsDEP1 and OsROC5. B, biallelic; M, monoallelic; W, wild type. b Sequences of individual mutated T1 rice plants at the target site. PAM is in red and crRNA in blue
LbCas12a is very sensitive to temperature in Arabidopsis
We next sought to generate heritable mutations in the dicot model plant, Arabidopsis, in which Cas12a-based mutagenesis has not been demonstrated. We decided to use a dual Pol II promoter for the LbCas12a system because it resulted in high mutation frequencies in rice [17] and its activity seems less sensitive to temperature (Fig. 1). The pZmUbi promoter used to express LbCas12a, and the crRNA here was also previously applied for Cas9 genome editing in Arabidopsis with high efficiency [31]. We targeted GL2 (GLABRA 2) and TT4 (TRANSPARENT TESTA 4) genes with one crRNA for each gene. The WT Arabidopsis plants (Col) were transformed with two corresponding T-DNA constructs via the floral dip method. The Arabidopsis plants were grown at 22 °C, a temperature at which LbCas12a displayed a good editing activity in rice (Fig. 1a, b). T1 transgenic plants were screened by RFLP, as mutations induced by LbCas12 would likely abolish the restriction enzyme site of SalI at both GL2 and TT4 target sites. However, we could not identify a single LbCas12a T1 plant that seemed to carry targeted mutations at either site (data not shown).
The surprising results prompted us to investigate whether LbCas12a requires a higher temperature to be active in Arabidopsis. To test this hypothesis, we decided to try 29 °C as a high temperature because Cas12a nucleases required a temperature of 28 °C or higher to reach optimal activity in rice (Fig. 1). We harvested seeds of T2 population from selected T1 lines. These T2 lines were grown on MS-hygromycin selection medium at 22 °C and 29 °C for 2 weeks, and then were used for DNA extraction followed by RFLP-based mutation analysis. For GL2, T2 line #8 showed mutations at 29 °C, but not at 22 °C (Fig. 4c). Further quantified data indicated a mutation frequency of 35% at 29 °C for T2 line #8 (Fig. 4b). Analysis of three additional GL2 T2 lines all revealed detectable mutation frequencies at 29 °C, but not at 22 °C (Fig. 4b). Similarly, while two examined T2 lines (#2 and #7) for TT4 showed mutation frequencies of about 14% and 15% at 29 °C, no mutation could be detected at 22 °C (Fig. 4c, d). We also tested a late treatment of transgenic plants at 29 °C (see the “Methods” section), which was less effective in inducing LbCas12a-mediated mutations at both target sites (Additional file 1: Figure S7). In addition, we compared LbCas12a and AsCas12a at editing the TT4 locus in protoplasts made from Arabidopsis rosette leaves. LbCas12a showed higher editing activity at 29 °C than at 22 °C (Additional file 1: Figure S8a). However, editing activity of AsCas12a was undetectable at this locus under both temperatures (Additional file 1: Figure S8b).

Activity of LbCas12a is highly sensitive to temperature in Arabidopsis somatic cells. Examples of RFLP gel of GL2 line #8 (a) and TT4 line #2 (c). Presence of a third band indicates mutations. Mutation percentages of GL2 lines #2, #3, #8, and #9 (b) and TT4 lines #2 and #7 (d; plants grown at 29 °C (blue) or 22 °C (red)). Error bars represent standard deviations of five biological replicates
Germline editing by LbCas12a in Arabidopsis with high-temperature treatment
We then developed a high-temperature treatment regime for GL2 and TT4 T2 plants with early treatment of 29 °C for about 4 weeks before moving the plants to 22 °C for recovery. T3 plants from these T2 parents were grown in a greenhouse at ~ 22 °C to demonstrate stable germline editing and evaluate mutation frequencies. Leaf tissues were collected from the three lines (GL2 #7-4, GL2 #7-7, and TT4 #9-7) for RFLP and Sanger sequencing (Fig. 5a, b). Thirty-three percent of the plants from GL2 #7-4, 21% of plants from GL2 #7-7, and 5.8% from TT4 #9-7 had targeted mutations (Fig. 5a). Among T3 lines from GL2 #7-4, the same mutation (a deletion of 5 bp) was found in all the homozygous and heterozygous plants, suggesting germline transmission from the parent (Fig. 5b). All homozygous GL2 #7-4 plants had a trichome-less (gl2 loss-of-function) phenotype (Fig. 5c). For GL2 #7-7 T3 plants, only GL2 #7-7-1 showed a loss-of-function phenotype. Sequencing analysis showed that the other homozygous or biallelic plants (e.g., GL2 #7-7-14 and GL2 #7-7-17) had the same allele of 3-bp deletion, suggesting the mutated protein, albeit missing one amino acid, is still functional. The LbCas12a reagent seems less efficient at the TT4 locus since only two heterozygous mutants were identified out of 34 TT4 #9-7 T3 lines (Fig. 5a, b). We followed two homozygous GL2 #7-4 lines to the T4 generation and found the mutations were germline transmitted (data not shown). Together, we demonstrated LbCas12a-mediated germline editing in Arabidopsis with a high-temperature treatment.

Germline mutagenesis by LbCas12a in Arabidopsis with high-temperature treatment. a A summary of genoty** results from GL2 #7-4, GL2 #7-7, and TT4 #9-7. Ho, homozygous; He, heterozygous; B, biallelic; W, wild type. b Target site sequences of gl2 mutants from parental line #7-4, #7-7, and sequences of tt4 mutants. c Images of gl2 mutant (GL2 #7-4-7) and wild type Arabidopsis (WT)
High-frequency genome editing by LbCas12a in maize at 28 °C
Our data in rice and Arabidopsis collectively suggest a temperature around 28 °C could be an optimal and practical temperature for achieving high-efficiency plant genome editing by Cas12a nucleases. To validate whether this is widely applicable to other plant species, we further tested high-temperature regimes in maize, an important commodity crop in which we recently demonstrated genome editing with LbCas12a [20]. We grew two independent T0 lines expressing LbCas12a and ZmGL2-crRNA1 (A842B-2-2 and A842B-5-1), along with the WT plants, in a greenhouse with a 16-h/8-h photoperiod and the temperature setting of 28 °C/21 °C (day/night). These T0 plants were crossed with WT B104 lines (the pollen donor) and kept in the same environment until maturity for harvesting T1 seeds (Fig. 6a). We genotyped T1 seedlings by Sanger sequencing (Fig. 6b). For A842B-2-2, 51.4% of T1 plants were mutated (18 out of 35). For A842B-5-1, 100% of T1 plants were mutated (32 out of 32), suggesting high mutagenesis frequencies. Biallelic mutants were obtained in T1 plants, and many new mutations were generated in the T1 generation (Fig. 6b and Additional file 1: Figure S9), indicating high activity of LbCas12a in maize at 28 °C.

High-temperature regime in maize T0 transgenic plants enables high-frequency mutagenesis by LbCas12a. a Diagram of maize transformation and heat treatment. Transgenic plants are crossed with a wild type inbred B104 pollen donor. B, biallelic; M, monoallelic; W, wild type. b Mutation rates and genoty** results of T1 generation from two maize mutant lines, A842B-2-2 and A842B-5-1
Probing Cas12a temperature sensitivity with a dLbCas12a-SRDX repressor
While the above-mentioned results clearly demonstrated activities of Cas12a nucleases are temperature sensitive, it is less clear which of the underlining mechanisms are affected. Before Cas12a can cleave DNA, it must scan the genome, find a PAM and crRNA complimentary sequence, unwind the DNA, and make conformational changes that activate nuclease activity [32, 33]. Temperature may affect any one, or all, of these steps. Although testing all these in vivo will be challenging, we reasoned that we could test whether Cas12a in planta binding to DNA is affected by temperature. Given the contrasting differences of LbCas12a editing activities in Arabidopsis at 22 °C and 29 °C, we decided to test this in Arabidopsis by using a dLbCas12a-SRDX repressor that we had previously developed [17]. We constructed a dLbCas12a-SRDX vector to target the promoter of PAP1 (PRODUCTION OF ANTHOCYANIN PIGMENT 1) with a single crRNA (Fig. 7a). Successful transcriptional repression would indicate successful binding of deactivated LbCas12a (dLbCas12a) to the target DNA. We selected two independent T1 lines for obtaining T2 generation seeds. T2 plants were germinated in MS-hygromycin selection medium for a week and then transplanted to new MS medium for 1-week treatment at 16 °C, 22 °C, and 29 °C, before analysis for gene expression by quantitative real-time (qRT)-PCR. The highest levels of repression were seen at 16 °C, where the expression of PAP1 was reduced to 30% and 20% of WT expression in the two transgenic lines, respectively (Fig. 7b). At 22 °C, expression of PAP1 was reduced to 40% of the WT in both lines (Fig. 7c). At 29 °C, PAP1 was also repressed, but more variation was seen between the two lines (Fig. 7d). Unsurprisingly, a comparative analysis of PAP1 in control plants shows that expression was higher at 29 °C, likely as a response to heat stress (Additional file 1: Figure S10). This could explain some variation between the lines and larger error bars for the repression results at 29 °C. Because repression was accomplished at lower temperatures, these results suggest that unlike genome editing, binding of LbCas12a to the target DNA is not abolished at lower temperatures. Further analysis of dLbCas12a-SRDX expression in transgenic lines indicated that its mRNA level was not elevated at a higher temperature (e.g., 29 °C) (Additional file 1: Figure S11). Rather, PAP1 #2 line showed higher expression of dLb-cas12a-SRDX at 16 °C (Additional file 1: Figure S11), which is consistent with stronger transcriptional repression on the target gene (PAP1) observed in this line at 16 °C (Fig. 7b).

Quantitative real-time (qRT)-PCR showing dLbCas12a-mediated transcriptional repression in Arabidopsis at different temperatures. a Diagram of a dCas12a-SRDX repressor targeting PAP1. Relative expression of PAP1 mRNA, normalized to EF1α, for two lines at 16 °C (b), 22 °C (c), and 29 °C (d). Error bars represent standard errors of four biological replicates from dCas12a-SRDX transgenic lines PAP #1 and #2, and three biological replicates from WT control plants
Discussion
In this study, we investigated temperature sensitivity of different Cas12a nucleases and applied high-temperature treatment for genome editing in plants. First, we found in rice cells AsCas12a is more sensitive to temperature than LbCas12a, and our results are consistent with the observation in zebrafish [29]. By doing stable rice transformation at a higher temperature, we showed that AsCas12a can induce heritable mutations at frequencies of 77.8% and 92.8% at two target sites. Hence, we not only demonstrated the use of AsCas12a for generating mutated plants for the first time, but also presented an efficient AsCas12a rice genome editing system, with mutagenesis frequencies close to those that we previously established for LbCas12a [17] and FnCas12a [18]. Interestingly, although we found mutation profiles for the three Cas12a nucleases were different from each other in rice cells, the mutation profiles for AsCas12a were much more different from those of FnCas12a and LbCas12a. While an in vitro DNA cleavage assay revealed AsCas12a, LbCas12a, and FnCas12a, all cleave approximately after the 18th base (relative to PAM) on the non-targeted strand and after the 23rd base on the targeted strand [3], genome-wide studies in human cells also suggested different mutation repair profiles of AsCas12a and LbCas12a [34, 35]. Further, both in vitro and in vivo data have suggested that AsCas12a has higher targeting specificity than LbCas12a [3, 34]. More recently, kinetic basis for high targeting specificity of AsCas12a was revealed [36]. These previous studies highlighted the importance of develo** AsCas12a-based genome editing systems, supporting the significance of our work on improving AsCas12a activity in plants. We hope our success with AsCas12a in rice will facilitate future research into applying and improving AsCas12a for genome editing in other plant species.
We also observed temperature sensitivities for LbCas12a in Arabidopsis. While LbCas12a showed reasonable nuclease activities in rice protoplasts and detectable activity in Arabidopsis protoplasts at 22 °C, it barely worked in Arabidopsis cells of stable transgenic lines at the same temperature. This could be due to the procedures of delivering CRISPR-Cas12a reagents, in vitro cell culture for the transient protoplast assay and floral dip for Arabidopsis. Also, the length of Cas12a treatment and the tissue sources for evaluating mutagenesis are different among transient and stable transgenesis. It is possible that any of these factors had contributed to the drastic difference between rice and Arabidopsis on editing efficiencies we observed. It is likely that chromatin structure plays a role as it has been shown to impact genome editing. For example, the natural cycle of nucleosome breathing and ATP-driven chromatin remodelers are essential for Cas9 binding at target sites [37, 38], and the same can be true for Cas12a. Recent comparative studies have revealed distinct chromatin packing in rice and Arabidopsis [39, 40]. It will be interesting to investigate how chromatin states in different plant species impact Cas12a and other CRISPR-Cas systems on genome editing. Nevertheless, we were able to rescue LbCas12a activity at a higher temperature and demonstrated LbCas12a-based germline editing in Arabidopsis. By contrast, AsCas12a showed undetectable editing activity at the TT4 locus in Arabidopsis protoplasts, which is consistent with the general observation in rice where AsCas12a has lower activity than LbCas12a. Since the baseline genome editing efficiency in Arabidopsis is much lower than that in rice, more future improvement is required in order to establish an efficient genome editing system in Arabidopsis using AsCas12a. Since we did not test the effects of temperature change on AsCas12a in rice seedlings, it is possible that using high temperature at different stages (e.g., seedlings vs calli) may result in different effects.
We also applied a high-temperature regime for achieving LbCas12a-based high-frequency genome editing in maize T1 lines. We showed that when transgenic maize lines expressing LbCas12a and crRNA were crossed to the WT plants, mutated T1 lines can be generated at a frequency as high as 100%, provided that the entire process of generating the T1 generation is done at a day time temperature of 28 °C. Many of the mutations discovered in the T1 lines are de novo generated new mutations. Our data were consistent with the recent report that new mutations could be generated in gametes or zygotes by Cas9 in maize [41]. Although we crossed our LbCas12a lines to WT, it is conceivable that we can also cross these transgenic lines to other transformation-recalcitrant maize varieties to knock out genes in different maize genetic backgrounds, as was shown previously with Cas9 [42]. Following our recent demonstration of LbCas12a in maize [20], the work here further streamlined an efficient LbCas12a system for targeted mutagenesis in maize.
In addition, we showed that Cas9 is temperature sensitive in rice, which is consistent with the recent report that investigated this issue in Arabidopsis and citrus [30]. Interestingly, Cas9 showed similar nuclease activities at 22 °C and 28 °C, and we started to see increased nuclease activity in rice protoplasts at 32 °C (Fig. 1c). Cas9 displayed optimal nuclease activity in human cells at 37 –39 °C [43], and a heat stress of 37 °C was applied for improving Cas9 editing in plants [30]. In zebrafish, AsCas12a had poor activity at 28 °C and its activity was drastically improved by elevating the temperature to 34 °C [29]. By contrast, we found Cas12a nucleases seem to reach optimal activities in plants at around 28–29 °C, which is more feasible given most plants grow in temperatures around 22–29 °C. For example, we have grown rice and maize constantly at 28 °C and have treated Arabidopsis at 29 °C for up to ~ 4 weeks continuously. However, lengthy and continuous treatment at 29 °C significantly impedes Arabidopsis growth. Further exploration of heat treatment regimens will probably result in more robust genome editing in plants. Hence, while Cas12a nucleases are temperature sensitive, it should not be a barrier that prevents adoption of them for genome editing in many other plant species.
A question remains, however, as why Cas12a-based genome editing is temperature sensitive. With the analysis of NHEJ mutations by all three Cas12a nucleases across four different temperatures, we ruled out the possible involvement of DNA repair pathways in this difference. Opposite effects of high temperatures on the activities of ZFN and TALEN versus CRISPR-Cas9 were reported in mammalian cells [43], which also suggested the effects were unlikely due to DNA repair machinery. Using a dLbCas12a-SRDX repressor, we further demonstrated that the DNA binding property of LbCas12a at lower temperatures is as good as, if not better than, higher temperatures. This is consistent with our previous observation that the dAsCas12a-SRDX repressor could work robustly in mediating targeted transcriptional repression at room temperature in Arabidopsis [17]. While DNA binding is not significantly affected under these temperatures, it is still possible that chromatin structure is affected by temperature in a way that impacts the necessary conformation change of the Cas12a/crRNA ribonucleoprotein complex that is required for activation of nuclease activity, as supported by the data in zebrafish [29]. Our data collectively points to a working hypothesis that Cas12a nuclease activity is affected by temperature. Upon activation, Cas12a proteins also unleash single-stranded DNase activities [44, 45]. We predict such non-specific DNase activities are likely also temperature sensitive. Finally, given that we have narrowed down the main cause of temperature sensitivity to Cas12 nuclease activities, it will be highly valuable and should also be possible to engineer Cas12a variants that are more active at lower temperatures, similar to engineering Cas12a variants with altered PAM specificities [25], which we and others have recently demonstrated in plants [18, 26].
Methods
T-DNA vector construction
T-DNA vectors for CRISPR-Cas9 were constructed based on the protocols described previously [46]. Briefly, forward and reverse oligos for OsPDS-gRNA03 (Additional file 1: Table S1) were phosphorylated, annealed, and ligated into pTX172 (Addgene #89259) at its BsaI sites.
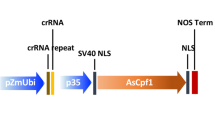
T-DNA vectors for CRISPR-Cas12a were constructed based on the protocols described previously [17]. Forward and reverse oligos for OsROC5-crRNA01, OsDEP1-crRNA02, AtGL2-crRNA1, and AtTT4-crRNA1 (Additional file 1: Table S1) were phosphorylated, annealed, and cloned into the Esp3I sites of pYPQ141-ZmUbi-RZ-As (Addgene #86196), pYPQ141-ZmUbi-RZ-Lb (Addgene #86197), and pYPQ141-ZmUbi-RZ-Fn (Addgene #108864). The resulting crRNA expression vectors were mixed with pYPQ203 (destination vector, Addgene #86207) and with pYPQ220 (AsCas12a, Addgene #86208), pYPQ230 (LbCas12a, Addgene #86210), and pYPQ239 (FnCas12a, Addgene #108859), to generate the final T-DNA binary vectors using multi-site LR reactions (1-5-2) [47, 48]. The LbCas12a-ZmGL2 T-DNA vector (A842B) for maize transformation was described previously [20].
The T-DNA vector for transcriptional repression in Arabidopsis was constructed similarly. The protospacer of AtPAP1-crRNA1 (Additional file 1: Table S1) was cloned into pYPQ141-ZmUbi-RZ-Lb at Esp3I sites in the form of phosphorylated and annealed oligos. Then, a multi-site LR reaction using pYPQ141-ZmUbi-RZ-Lb-AtPAP1-crRNA1, pYPQ233 (dLbCas12a-SRDX, Addgene #86211), and pYPQ202 (Addgene #86198) was conducted to generate the final T-DNA vector.
Rice protoplast transfection and stable transformation
The Japonica rice cultivar Nipponbare was used in this study. Polyethylene glycol (PEG)-mediated transfection of rice mesophyll protoplasts with T-DNA vectors was carried out according to our previously published protocol [46, 49]. After transfection, rice protoplasts were divided and incubated for 2 days at four temperatures, 22 °C, 28 °C, 32 °C, and 37 °C. A DNA construct carrying GFP marker gene was used as a control to determine the transfection efficiencies. Cells with green fluorescence were counted 2 days after the transfection.
Rice stable transformation was conducted as published previously [46]. Plants were grown in a growth chamber at 25 °C for co-culture, 32 °C for selection, and 28 °C for shoot generation.
Mutagenesis analysis at target sites
Genomic DNA was extracted from transfected rice protoplasts or leaves of transgenic lines using the cetyl trimethylammonium bromide (CTAB) method [50]. The genomic region flanking target sites were PCR amplified with primers DEP1-F 5′-TCACCGATTCTTTCCATGCG-3′, DEP1-R 5′-GCCACAATCGGGTTTGCATT-3′, ROC5-F 5′ CTTATGTTCCGTTCCAATCCT-3′, ROC5-R 5′ CCTACACTTCACATTTCCACCT-3′ and PDS-F 5′ GCTCACACTGTTTTGTCGTCC-3′, and PDS-R 5′ ATCATATGCAGCGCTGGAGT-3′. DEP1 PCR products were digested with BglII and ROC5 PCR products were digested with NlaIII for RFLP analysis. PDS PCR products were digested with AseI. The PCR products were digested by their corresponding restriction enzymes overnight and then analyzed by agarose gel electrophoresis. Mutations in T0 plants were further identified by Sanger sequencing of the PCR products.
High-throughput sequencing analysis
High-throughput sequencing analysis was conducted for transfected rice protoplast samples as published previously [46, 51]. The genomic region flanking target sites were PCR amplified using barcoded primers. Purified DNA samples were quantified by Qubit 2.0 Fluorometer (Life Technologies) and were sequenced using Illumina HiSeq 2500 platform. Data processing was carried out using CRISPRMatch [52].
Arabidopsis stable transformation, temperature treatment, and mutation analysis
Arabidopsis WT plants (Col) were transformed by the floral dip method [53]. Seeds for T1, T2, and T3 generations were sterilized using 50% bleach and 0.05% Tween, vernalized at 4 °C for 3 days, then plated to MS-hygromycin plates. After a week, transgenic plants were transferred to MS clean plates for a week of recovery before soil transplantation. To test a variety of T2 lines, five individual plants were sacrificed after 2 weeks of heat treatment at 29 °C on MS plates. Leaf tissue was used for DNA extraction using a modified CTAB method [50]. The rest of the plants were transferred to soil and kept at 29 °C for a total of 29 days before recovery at 22 °C. A second batch of plants was heat treated to test an alternative method of late treatment at 29 °C. For this, 6 days after plating, the plants were kept at 29 °C for 8 days. After 24 days of recovery at 22 °C, plants were kept at 29 °C again for 14 days.
For mutation analysis, a ~ 677 bp fragment covering the GL2 target site was amplified using the primers GL1-F1 5′-GATGGCTGCCAATGCTGTAGCTGG-3′ and GL2-R1 5′-CGTCAACTACTCTTCTGCCCAGG-3′, and a ~ 400-bp fragment covering the TT4 target site was amplified using the primers TT4-F2 5′-AGGCATCTTGGCTATTGGCACTG-3′ and TT4-gR3-top 5′-gattGGGCTGGCCCCACTCCTTGA-3′. GL2 and TT4 PCR products underwent RFLP analysis through direct digestion with restriction enzyme SalI and analysis on 1.5% and 2% agarose gels, respectively. The mutation percentages for each plant were estimated from RFLP gels with Image Lab (BioRad). PCR products were cleaned using Exonuclease I and Antarctic Phosphatase and sent for Sanger sequencing. Results were aligned in Snapgene and decoded with CRISP-ID (GSL Biotech LLC) [54].
Arabidopsis protoplasts isolation and transformation
Arabidopsis protoplast isolation and transformation were based on our previous study [49, 55]. Arabidopsis Col-0 plants were grown under 12-h light/12-h dark photoperiod at 22–25 °C for 4 weeks. About 10–20 fresh leaves were cut into leaf strips by razor blades at 0.5–1 mm in width. Leaf strips were quickly transferred into 8–10 ml enzyme solution (1.0–1.5% Cellulase R10, 0.25% Macerozyme R10, 0.4 M Mannitol, 20 mM MES, 20 mM KCl, 10 mM CaCl2, 0.1% BSA, and pH 5.8), followed by vacuum infiltration for 30 min. The leaf strips were digested by shaking at 30 rpm for 1–4 h at 25 °C in the dark. The digested products were filtered with 70-μm nylon mesh into 50-ml tube with 10 ml W5 buffer. The protoplasts were collected at 200×g for 2 min. Then, they were resuspended with 10 ml W5 buffer twice and were centrifuge at 100×g for 1 min at RT. The cells were suspended in MMG buffer (0.4 M Mannitol, 4 mM MES, 15 mM MgCl2, pH 5.8) for 1 × 105 cells/ml. Two hundred microliter protoplasts were mixed with 30 μl plasmid (30 μg) and 230 μl PEG buffer (40% w/v PEG4000, 0.2 M mannitol, and 0.1 M CaCl2) for incubation at 23 °C for 30 min. After adding 900 μl W5 buffer to stop transformation, the protoplasts were centrifugal at 200×g for 5 min and resuspended in 1 ml WI buffer (0.5 M mannitol, 20 mM KCl, and 4 mM MES at pH 5.7), and then transferred into two six-well culture plates in equal amount and incubated at 22 °C and 29 °C. After incubation for 24 h, the protoplasts were collected for DNA extraction and further analysis.
Transgenic T1 maize seeds, temperature treatment, and mutation analysis
Transgenic maize T1 seeds originated from two maternal T0 lines carrying a LbCas12a construct and on-target mutations in the target gene GL2 (A842B-2-2 and A842B-5-1) [20]. These maternal T0 lines had been crossed with wild-type B104 pollen donors. The T0 lines were grown in a greenhouse with a 16-h/8-h photoperiod, and the temperature setting of 28 °C/21 °C (day/night). For each T0 line, 20 T1 seeds were sown on a tray containing Metro-Mix 900 potting mix (Sun-Gro Horticulture, Agawam, MA). Leaf tissues, first and second leaves, were collected from each T1 plant and pooled for genomic DNA isolation. The presence of T-DNA was confirmed by PCR using the primers LbCas12a-F 5′-AATGGAACGCGGAGTATGAC-3′ and LbCas12a-R 5′-ACATGTCGCCCTTCTTGAAC-3′ [20]. The genoty** of the T1 plants were performed by PCR and sequencing using the primers Zm-gl2-F2 5′-CACAGCCTTGCAATCAATTC-3′, Zm-gl2-R2 5′-GCTGACGTGGAAGGAGTAGC-3′, and ZmGl2-exon2-F1 5′-ACACCGTGTCTTCGTCAAAA-3′ [20]. About 1 kb fragment flanking, the LbCas12a target site was amplified using the oligonucleotides Zm-gl2-F2 and Zm-gl2-R2, and single-band amplification was confirmed by 1% agarose gel electrophoresis. PCR products were cleaned up by ExoSAP-IT kit (ThermoFisher Scientific) according to the manufacturer’s instruction. The oligonucleotide ZmGl2-exon2-F1 was used for Sanger sequencing, and the resulting trace files were analyzed by the Tracking of Indels by Decomposition (TIDE) [56] and DSDecode [57].
Transcriptional repression in Arabidopsis
Arabidopsis dLbCas12a-PAP1 T2 plants were grown on MS media with hygromycin at 22 °C for a week. They were then transferred to MS medium without hygromycin, allowed to recover at 22 °C for 2 days, and then grown at 16 °C, 22 °C, and 29 °C for a week. Arabidopsis leaf tissue was collected from these T2 seedlings. The qRT-PCR analysis was carried out following previously described protocols [47] with minor modifications. GUS transformed plants, also expressing a hygromycin resistance gene, were used as controls. Total RNA was extracted using TRIzol™ Reagent (Invitrogen) following the manufacturer’s instructions with the exception that samples were extracted twice using chloroform and washed twice by 75% ethanol. RNA was treated with DNase I (New England BioLabs) to remove DNA contamination. Complementary DNA (cDNA) was synthesized using SuperScript III First-Strand Synthesis System (Invitrogen) with Oligo dT. The qRT-PCR was set up using Applied Biosystems SYBR Green Master Mix (Invitrogen) and ran on the CFX96 Touch™ Real-Time PCR Detection System. The following primers were used for the transgenic plant samples: PAP1-F 5′-AGTATGGAGAAGGCAAATGGC-3′ and PAP1-R 5′-CACCTATTCCCTAGAAGCCTATG-3′, LbCas12a-RT-F1 5′-TTCGTTCAACGGATTCACAA-3′, and LbCas12a-RT-R1 5′-GCTTGTCAAAAATTGCGTCA-3′. Elongation factor 1 α (EF1α) was used as the internal control and amplified with the following primers: EF-1α-F 5′-TGAGCACGCTCTTCTTGCTTTCA-3′ and EF-1α-R 5′-GGTGGTGGCATCCATCTTGTTACA-3′. The average of three technical replicates was used for data analysis of each biological replicate. Relative expression to controls was calculated using the comparative threshold cycle method.
References
Voytas DF. Plant genome engineering with sequence-specific nucleases. Annu Rev Plant Biol. 2013. https://doi.org/10.1146/annurev-arplant-042811-105552.
**ek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012. https://doi.org/10.1126/science.1225829.
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015. https://doi.org/10.1016/j.cell.2015.09.038.
Ma X, Zhu Q, Chen Y, Liu Y-G. CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol Plant. 2016. https://doi.org/10.1016/j.molp.2016.04.009.
Malzahn A, Lowder L, Qi Y. Plant genome editing with TALEN and CRISPR. Cell Biosci. 2017. https://doi.org/10.1186/s13578-017-0148-4.
Murovec J, Pirc Ž, Yang B. New variants of CRISPR RNA-guided genome editing enzymes. Plant Biotechnol J. 2017. https://doi.org/10.1111/pbi.12736.
Yin K, Gao C, Qiu J-L. Progress and prospects in plant genome editing. Nature Plants. 2017. https://doi.org/10.1038/nplants.2017.107.
Schindele P, Wolter F, Puchta H. Transforming plant biology and breeding with CRISPR/Cas9, Cas12 and Cas13. FEBS Lett. 2018. https://doi.org/10.1002/1873-3468.13073.
Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016. https://doi.org/10.1038/nature17945.
Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, et al. Multiplex gene editing by CRISPR-Cpf1 through autonomous processing of a single crRNA array. Nat Biotechnol. 2017. https://doi.org/10.1038/nbt.3737.
Swarts DC, van der Oost J, **ek M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol Cell. 2017. https://doi.org/10.1016/j.molcel.2017.03.016.
Wang M, Mao Y, Lu Y, Tao X, Zhu J-K. Multiplex gene editing in rice using the CRISPR-Cpf1 system. Mol Plant. 2017. https://doi.org/10.1016/j.molp.2017.03.001.
Ferenczi A, Pyott DE, **pnitou A, Molnar A. Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. PNAS. 2017. https://doi.org/10.1073/pnas.1710597114.
Endo A, Masafumi M, Kaya H, Toki S. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep. 2016. https://doi.org/10.1038/srep38169.
Hu X, Wang C, Liu Q, Fu Y, Wang K. Targeted mutagenesis in rice using CRISPR-Cpf1 system. J Genet Genomics. 2017. https://doi.org/10.1016/j.jgg.2016.12.001.
Xu R, Qin R, Li H, Li D, Li L, Wei P, et al. Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol J. 2017. https://doi.org/10.1111/pbi.12669.
Tang X, Lowder LG, Zhang T, Malzahn AA, Zheng X, Voytas DF, et al. A CRISPR–Cpf1 system for efficient genome editing and transcriptional repression in plants. Nature Plants. 2017. https://doi.org/10.1038/nplants.2017.103.
Zhong Z, Zhang Y, You Q, Tang X, Ren Q, Liu S, et al. Plant genome editing using FnCpf1 and LbCpf1 nucleases at redefined and altered PAM sites. Mol Plant. 2018. https://doi.org/10.1016/j.molp.2018.03.008.
Kim H, Kim S-T, Ryu J, Kang B-C, Kim J-S, Kim S-G. CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun. 2017. https://doi.org/10.1038/ncomms14406.
Lee K, Zhang Y, Kleinstiver BP, Guo JA, Aryee MJ, Miller J, et al. Activities and specificities of CRISPR-Cas9 and Cas12a nucleases for targeted mutagenesis in maize. Plant Biotechnol J. 2018. https://doi.org/10.1111/pbi.12982.
Tang X, Liu G, Zhou J, Ren Q, You Q, Tain L, et al. A large-scale whole-genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1(Cas12a) nucleases in rice. Genome Biol. 2018. https://doi.org/10.1186/s13059-018-1458-5.
Kim HK, Song M, Lee J, Menon AV, Jung S, Kang Y-M, et al. In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nat Methods. 2017. https://doi.org/10.1038/nmeth.4104.
Zhu H, Liang C. CRISPR-DT: designing gRNAs for the CRISPR-Cpf1 system with improved target efficiency and specificity. In: bioRxiv; 2018. https://doi.org/10.1101/269910.
Tu M, Lin L, Cheng Y, He X, Sun H, ** of targeted mutations. Mol Plant. 2015. https://doi.org/10.1016/j.molp.2015.05.009.
Acknowledgements
T0 transgenic maize lines were produced by Iowa State University Plant Transformation Facility.
Funding
This work was supported by Syngenta, the National Science Foundation (IOS-1758745), Foundation for Food and Agriculture Research (593603), and startup funds provided by University of Maryland to YQ. This work was also supported by National Science Foundation of China (31771486) and the Sichuan Youth Science and Technology Foundation (2017JQ0005) and the Science Strength Promotion Program of UESTC to YZ, by the National Transgenic Major Project (2018ZX08020-003) and the Open Foundation of Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding (PL201801) to YZ, and TZ, the Jiangsu Specially-Appointed Professor and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) to TZ, and by the BRAG grant (2016–06247) to KW.
Availability of data and materials
The raw data of deep sequencing have been deposited to the Genome Sequence Archive in Bei**g Institute of Genomics (BIG) under the accession number PRJCA000992 (http://bigd.big.ac.cn/bioproject/browse/PRJCA000992) and the Sequence Read Archive in National Center for Biotechnology Information (NCBI) under the accession number SRP158345 (https://www.ncbi.nlm.nih.gov/sra/SRP158345). Raw data for Figs. 1, 2, 4, 7 and Additional file 1: Figure S3, S4, S5, S8, S10, S11 can be found in ‘Additional file 2: Raw data’.
Author information
Authors and Affiliations
Contributions
YQ conceived the project. YQ, Yong Z, KW, AM, XT, and KL designed the experiments. AM, XT, KL, QR, SS, Yingxiao Z, HC, MK, XZ, KD, and VS conducted the experiments. AM, XT, KL, SS, HC, YB, TZ, KW, Yong Z, and YQ analyzed the data. AM, Yong Z, and YQ wrote the manuscript draft. All authors participated in the discussion and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1.RFLP analysis of nuclease activities of three Cas12a (Cpf1) nucleases in rice protoplasts under different temperatures. Figure S2. RFLP analysis of SpCas9 activity in rice protoplasts under different temperatures. Figure S3. Measurement of rice protoplast transfection efficiencies using GFP construct at different temperatures. Figure S4. Three Cas12a nucleases generate slightly distinct mutation profiles at OsROC5 site which are unaffected by temperature. Figure S5. Mutation profiles by SpCas9 are largely unaffected by temperature. Figure S6. RFLP analysis of T0 mutant lines by AsCas12a. Figure S7. Late treatment of high temperature is ineffective on promoting LbCas12a-induced mutations in Arabidopsis. Figure S8. Analysis of AsCas12a-mediated mutagenesis at the TT4 locus in Arabidopsis protoplasts. Figure S9. Genoty** results of T1 maize lines. Figure S10. PAP1 expression at three different temperatures in the GUS control plants (WT). Figure S11. dCas12a-SRDX expression at three different temperatures in transgenic lines. Table S1. Guide RNA oligos. (PPTX 10187 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Malzahn, A.A., Tang, X., Lee, K. et al. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol 17, 9 (2019). https://doi.org/10.1186/s12915-019-0629-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-019-0629-5