
Abstract
Background
Biallelic pathogenic variants in PIP5K1C (MIM #606,102) lead to lethal congenital contractural syndrome 3 (LCCS3, MIM #611,369), a rare autosomal recessive genetic disorder characterized by small gestational age, severe multiple joint contractures and muscle atrophy, early death due to respiratory failure. Currently, 5 individuals with LCCS3 were reported and 5 pathogenic variants in PIP5K1C were identified. Here, we reported the two fetuses in a Chinese pedigree who displayed multiple joint contractures and other congenital anomalies.
Methods
Trio-based whole-exome sequencing (WES) was performed for the parents and the recent fetus to detect the genetic cause for fetus phenotype.
Results
A novel variant, NM_012398.3: c.949_952dup, p.S318Ifs*28 and a previously reported variant, c.688_689del, p.G230Qfs*114 (ClinVar database) in PIP5K1C, were detected in the individuals, and these variants were inherited from the mother and father, respectively. We described the features of multiple joint contractures in our fetuses, including bilateral talipes equinovarus, stiffness in the limbs, extended knees, persistently closed hands and overlap** fingers, which have not been delineated detailedly in previously reported LCCS3 individuals. Furthermore, novel phenotype, bilateral dilated lateral ventricles, was revealed in one fetus.
Conclusions
These findings expanded the genetic variant spectrum of PIP5K1C and enriched the clinical features of LCCS3, which will help with the prenatal diagnosis and genetic counseling for this family.
Similar content being viewed by others


Background
Lethal congenital contracture syndrome (LCCS) is a heterogenous group of congenital genetic condition. It is characterized by multiple joint contractures, polyhydramnios and reduced fetal movement, often leading to the perinatal or neonatal death. Studies have suggested that LCCS is an autosomal recessive disorder causally linked to 11 genes, including GLE1, ERBB3, PIP5K1C, MYBPC1, DNM2, ZBTB42, CNTNAP1, ADCY6, ADGRG6, NEK9 and GLDN. Among them, LCCS1 (MIM #253,310), LCCS7 (MIM #607,598) and LCCS11 (MIM #617,194) have been relatively frequent reported in the literature [1,2,3,4,5,6]. However, lethal congenital contracture syndrome 3 (LCCS3 MIM #611,369), caused by biallelic pathogenic variants in PIP5K1C (MIM #606,102), was rarely reported.
The PIP5K1C gene consists of 18 exons and encodes a 668-amino acid enzyme. This protein utilizes phosphatidylinositol 4-phosphate (PI4P) as a substrate to synthesize phosphatidylinositol 4,5-phosphate (PIP2) on the cell membrane [7, 1B, II-2). Then, she underwent vaginal delivery prematurely at 26 weeks, and the baby passed away after birth due to respiratory failure. In the recent pregnancy, the woman sought medical attention due to advanced maternal age and progressively reduced fetal movement. At 23 weeks of gestation, a prenatal ultrasound scan revealed bilateral dilated lateral ventricles (13.4 mm). Additionally, the fetus exhibited stiffness in the limbs, extended knees, bilateral talipes equinovarus and persistently closed hands (Fig. 1B, II-4). The karyotype analysis and chromosomal microarray of the amniotic fluid were normal. MLPA detected no deletion of exons 7 and 8 in SMN1.

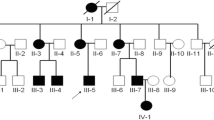
The pedigree chart and radiographic findings for the two fetuses. (A). Pedigree for a consanguineous Chinese family with LCCS3 (The black arrow represents the proband, translucent grey indicates carrier). (B). Radiographic findings for the two fetuses. (II-2) bilateral talipes equinovarus (a), flexion contractures of fingers and overlap** fingers (b). (II-4) bilateral dilated lateral ventricles (13.4 mm) (c), stiffness in the limbs, extended knees (d), bilateral talipes equinovarus (e) and persistently closed hands (f)
Genetic analysis
Compound heterozygous variants, NM_012398.3: c.949_952dup (p.S318Ifs*28) and c.688_689del (p.G230Qfs*114) in PIP5K1C, were revealed in II-4, which were inherited from the mother and father. Next, we analyzed the DNA sample of II-2, and it expectedly revealed the same PIP5K1C compound heterozygous variants. Sanger sequencing verification was performed for other members in the pedigree. Individual II-1 was proven to be wildtype and individual II-3 was an asymptomatic carrier (c.949_952dup, p.S318Ifs*28 in PIP5K1C). The variant segregates as autosomal recessive (Fig. 2). The paternally inherited frameshift variant, (c.688_689del, p.G230Qfs*114), has been reported in ClinVar database. The maternally inherited novel variant, c.949_952dup (p.S318Ifs*28), was predicted to cause protein truncation and was unlikely escape nonsense-mediated mRNA decay. In addition, the individuals’ phenotypes were highly consistent with that of LCCS3. Trio-based WES also excluded other possible known genetic causes. Thus, both variants were categorized as clinically pathogenic according to the ACMG/AMP guidelines. (PVS1 + PM2 + PP1 + PP4).

Variant confirmation by Sanger sequencing. Compound heterozygous variants NM_012398.3: c.688_689del and c.949_952dup in PIP5K1C were detected in both fetuses and their asymptomatic parents and siblings. The red arrow indicates the variant site
Discussion
PIP5K1C is mainly highly expressed in brain and plays an important role in neural signaling pathway [12, 13]. Pip5k1c -/- mice caused a 50% reduction in PIP2 in brain, leading to an impairment of its depolarization-dependent synthesis in nerve terminals and synaptic defects [14]. PIP5K1C has been demonstrated to regulate various cellular processes including receptor-mediated calcium signaling transmission, actin cytoskeleton dynamics, endocytosis and exocytosis [15]. Additionally, PIP5K1C plays a crucial role in the maintenance of bone development. It exerts its influence on bone growth and development by regulating the movement of calcium ions in cells and body fluids [18, 19], here our two fetuses also exhibited bilateral talipes equinovarus. Ankylosis of knee joint was observed in individuals with LCCS6, 7 and 9, here our fetus 2 (II-4) showed this feature [5, 18, 20]. Flexion contractures of fingers were reported in individuals with LCCS7, 9, 10, 11, which was also observed in our two fetuses [5, 18,19,20,21]. Our findings profiled the picture of multiple joint contractures in LCCS3. Polyhydramnios was a marked feature of LCCS [16,17,18,19,20,21,22,23,24,25]. However, this feature has not yet been observed in individuals with LCCS3, which deserves further investigation.
In conclusion, we described in detail the prenatal clinical features of a Chinese pedigree with LCCS3 caused by biallelic pathogenic variants in PIP5K1C. The identification of the novel variant and novel phenotypes expands the variant spectrum of PIP5K1C and enriches the clinical characteristics of LCCS3, which will be valuable for prenatal diagnosis and genetic counseling.
Data availability
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
References
Stals KL, Wakeling M, Baptista J, Caswell R, Parrish A, Rankin J, et al. Diagnosis of lethal or prenatal-onset autosomal recessive disorders by parental exome sequencing. Prenat Diagn. 2018;38(1):33–43. https://doi.org/10.1002/pd.5175.
Laquerriere A, Jaber D, Abiusi E, Maluenda J, Mejlachowicz D, Vivanti A, et al. Phenotypic spectrum and genomics of undiagnosed arthrogryposis multiplex congenita. J Med Genet. 2022;59(6):559–67. https://doi.org/10.1136/jmedgenet-2020-107595.
Yates TM, Campeau PM, Ghoumid J, Kibaek M, Larsen MJ, Smol T, et al. Biallelic variants in GLE1 with survival beyond neonatal period. Clin Genet. 2020;98(6):622–5. https://doi.org/10.1111/cge.13841.
Vanderver A, Simons C, Helman G, Crawford J, Wolf NI, Bernard G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. 2016;79(6):1031–7. https://doi.org/10.1002/ana.24650.
Laquérriere A, Maluenda J, Camus A, Fontenas L, Dieterich K, Nolent F, et al. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum Mol Genet. 2014;23(9):2279–89. https://doi.org/10.1093/hmg/ddt618.
Pergande M, Motameny S, Özdemir Ö, Kreutzer M, Wang H, Daimagüler HS, et al. The genomic and clinical landscape of fetal akinesia. Genet Med. 2020;22(3):511–23. https://doi.org/10.1038/s41436-019-0680-1.
Xu W, ** M, Huang W, Wang H, Hu R, Li J, Cao Y, Apical. PtdIns(4,5)P(2) is required for ciliogenesis and suppression of polycystic kidney disease. FASEB J. 2019;33(2):2848–57. https://doi.org/10.1096/fj.201800385RRR.
Yan Q, Gao H, Yao Q, Ling K, **ao G. Loss of phosphatidylinositol-4-phosphate 5-kinase type-1 gamma (Pip5k1c) in mesenchymal stem cells leads to osteopenia by impairing bone remodeling. J Biol Chem. 2022;298(3):101639. https://doi.org/10.1016/j.jbc.2022.101639.
Narkis G, Ofir R, Landau D, Manor E, Volokita M, Hershkowitz R, et al. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinsitol pathway. Am J Hum Genet. 2007;81(3):530–9. https://doi.org/10.1086/520771.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. https://doi.org/10.1038/gim.2015.148.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Legate KR, Montag D, Bottcher RT, Takahashi S, Fassler R. Comparative phenotypic analysis of the two major splice isoforms of phosphatidylinositol phosphate kinase type igamma in vivo. J Cell Sci. 2012;125(23):5636–46. https://doi.org/10.1242/jcs.102145.
White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, et al. Genome-wide generation and systematic phenoty** of knockout mice reveals new roles for many genes. Cell. 2013;154(2):452–64. https://doi.org/10.1016/j.cell.2013.06.022.
Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, et al. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431(7007):415–22. https://doi.org/10.1038/nature02896.
Yu YL, Chou RH, Chen LT, Shyu WC, Hsieh SC, Wu CS, et al. EZH2 regulates neuronal differentiation of mesenchymal stem cells through PIP5K1C-dependent calcium signaling. J Biol Chem. 2011;286(11):9657–67. https://doi.org/10.1074/jbc.M110.185124.
Lakhani S, Doan R, Almureikhi M, Partlow JN, Al Saffar M, Elsaid MF, et al. Identification of a novel CNTNAP1 mutation causing arthrogryposis multiplex congenita with cerebral and cerebellar atrophy. Eur J Med Genet. 2017;60(5):245–9. https://doi.org/10.1016/j.ejmg.2017.02.006.
Hosseini M, Fattahi Z, Abedini SS, Hu H, Ropers HH, Kalscheuer VM, et al. GPR126: a novel candidate gene implicated in autosomal recessive intellectual disability. Am J Med Genet A. 2019;179(1):13–9.
Ravenscroft G, Nolent F, Rajagopalan S, Meireles AM, Paavola KJ, Gaillard D, et al. Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita. Am J Hum Genet. 2015;96(6):955–61. https://doi.org/10.1016/j.ajhg.2015.04.014.
Casey JP, Brennan K, Scheidel N, McGettigan P, Lavin PT, Murphy H, et al. Recessive NEK9 mutation causes a lethal skeletal dysplasia with evidence of cell cycle and ciliary defects. Hum Mol Genet. 2016;25(9):1824–35. https://doi.org/10.1093/hmg/ddw054.
Patel N, Smith LL, Faqeih E, Mohamed J, Gupta VA, Alkuraya FS. ZBTB42 mutation defines a novel lethal congenital contracture syndrome (LCCS6). Hum Mol Genet. 2014;23(24):6584–93. https://doi.org/10.1093/hmg/ddu384.
Maluenda J, Manso C, Quevarec L, Vivanti A, Marguet F, Gonzales M, et al. Mutations in GLDN, Encoding Gliomedin, a critical component of the nodes of Ranvier, are responsible for Lethal Arthrogryposis. Am J Hum Genet. 2016;99(4):928–33. https://doi.org/10.1016/j.ajhg.2016.07.021.
Landau D, Mishori-Dery A, Hershkovitz R, Narkis G, Elbedour K, Carmi R. A new autosomal recessive congenital contractural syndrome in an Israeli Bedouin kindred. Am J Med Genet A. 2003;117A(1):37–40. https://doi.org/10.1002/ajmg.a.10894.
Koutsopoulos OS, Kretz C, Weller CM, Roux A, Mojzisova H, Bohm J, et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet. 2013;21(6):637–42. https://doi.org/10.1038/ejhg.2012.226.
Markus B, Narkis G, Landau D, Birk RZ, Cohen I, Birk OS. Autosomal recessive lethal congenital contractural syndrome type 4 (LCCS4) caused by a mutation in MYBPC1. Hum Mutat. 2012;33(10):1435–8. https://doi.org/10.1002/humu.22122.
Nousiainen HO, Kestila M, Pakkasjarvi N, Honkala H, Kuure S, Tallila J, et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40(2):155–7. https://doi.org/10.1038/ng.2007.65.
Acknowledgements
We would like to express our sincere gratitude to the pedigree for their cooperation. Furthermore, we extend our thanks to all the colleagues who actively participated in this study.
Funding
This study was supported by the Dongguan Social Development Project (No. 20221800900532 to Jianhua Luo). The funding body participated in the design of the project and interpretation of whole exome sequencing.
Author information
Authors and Affiliations
Contributions
FZ drafted the first versions of the manuscript. HMY and JHL were responsible for the design of the project, data analysis, and revised the manuscript. HMG, ZXD, QHX made the clinical evaluation and collected clinical information of the patients in detail. XLZ provided financial support. QMW performed the experiments and data entry.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by ethics committee of Dongguan Maternal and Child Health Hospital. Written informed consent was obtained from the patient’s parents for publication.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, F., Guo, H., Zhou, X. et al. Novel PIP5K1C variant identified in a Chinese pedigree with lethal congenital contractural syndrome 3. BMC Pediatr 24, 182 (2024). https://doi.org/10.1186/s12887-024-04674-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04674-6