
Abstract
Background
TBX6, a member of the T-box gene family, encodes the transcription factor box 6 that is critical for somite segmentation in vertebrates. It is known that the compound heterozygosity of disruptive variants in trans with a common hypomorphic risk haplotype (T-C-A) in the TBX6 gene contribute to 10% of congenital scoliosis (CS) cases. The deletion of chromosome 17q12 is a rare cytogenetic abnormality, which often leads to renal cysts and diabetes mellitus. However, the affected individuals often exhibit clinical heterogeneity and incomplete penetrance.
Methods
We here present a Chinese fetus who was shown to have CS by ultrasound examination at 17 weeks of gestation. Trio whole-exome sequencing (WES) was performed to investigate the underlying genetic defects of the fetus. In vitro functional experiments, including western-blotting and luciferase transactivation assay, were performed to determine the pathogenicity of the novel variant of TBX6.
Results
WES revealed the fetus harbored a compound heterozygous variant of c.338_340del (p.Ile113del) and the common hypomorphic risk haplotype of the TBX6 gene. In vitro functional study showed the p.Ile113del variant had no impact on TBX6 expression, but almost led to complete loss of its transcriptional activity. In addition, we identified a 1.85 Mb deletion on 17q12 region in the fetus and the mother. Though there is currently no clinical phenotype associated with this copy number variation in the fetus, it can explain multiple renal cysts in the pregnant woman.
Conclusions
This study is the first to report a Chinese fetus with a single amino acid deletion variant and a T-C-A haplotype of TBX6. The clinical heterogeneity of 17q12 microdeletion poses significant challenges for prenatal genetic counseling. Our results once again suggest the complexity of prenatal genetic diagnosis.
Similar content being viewed by others


Introduction
Congenital scoliosis (CS) is characterized by the lateral curvature of the spine resulting from vertebral anomalies that arise during intrauterine somitogenesis [1]. The estimated incidence of CS is 0.5–1/1,000 live births [2]. Highly synchronized developmental molecular pathways are involved in somitogenesis, with the Notch signal transduction pathway being crucial during early embryogenesis. The pathogenic variants of genes (MESP2, DLL3, LFNG, and HES7) in the Notch signaling pathway have been implicated in spondylocostal dysostosis (SCD) [3].
The transcription factor box 6 (TBX6), a member of the phylogenetically conserved T-box gene family, encodes transcription factors known for their highly conserved N-terminal DNA-binding domain (T-box), which plays a critical role in somite segmentation in vertebrates. Approximately 10% of CS cases can be attributed to the compound heterozygosity of null mutations (such as a deletion copy number variation (CNV) or a loss-of-function variant allele) in trans with a common hypomorphic risk haplotype (T-C-A) [Whole-exome sequencing and Sanger sequencing The karyoty** and CMA of fetal samples were conducted as described previously [11]. For trio-WES, genomic DNA was extracted from amniotic fluid and peripheral blood samples of the parents using the MagPure Buffy Coat DNA Midi KF Kit (MAGEN) according to the manufacturer’s instructions. 300 ng of DNA underwent shearing to produce 100–500 bp fragments. Fragments of 200–300 bp were selectively retained using VAHTS DNA clean beads (Vazyme, Nan**g, Jiangsu, China). The sample library was subjected to hybridization with customized gene fragment capture probes (KAPA HyperExome, Roche) according to the manufacturer’s instructions. Subsequently, the captured library was sequenced on the MGISEQ-2000 platform (MGI, Shenzhen, Guangdong, China) in accordance with the manufacturer’s instructions. The sequencing quality of the raw data was evaluated using the SOAPnuke software, with the removal of low-quality and contaminated reads to obtain clean reads. The clean reads were aligned to the GRCh37/hg19 human reference sequence using the Burrows-Wheeler Aligner. Single nucleotide variants, insertions, and deletions were identified using the GATK software package. The annotation and screening of variants were performed using the BGI-Varanno algorithm. The ExomeDepth software was used for exon CNV detection, while the CNV kit software was used for the CNV detection of large segments exceeding 1 M. Additionally, exon 3 of TBX6 and rs2289292, rs3809624, and rs3809627 were amplified via polymerase chain reaction from genomic DNA (primer sequences available upon request). Sanger sequencing was performed on an ABI 3700 sequencer (Applied Biosystems, Foster City, CA, USA). Conservation analysis of TBX6‑Ile113 was performed using Sequence Manipulation Suite: Color Align Properties (http://www.bioinformatics.org/sms2/color_align_prop.html). Since TBX6 crystal structure was not available, the three-dimensional (3D) structure of the wild-type (WT) TBX6 protein was simulated using Pymol v.1.8.4.0 software (https://www.pymol. org) using the amino acid sequence of TBX3 (NP_005987.3). Wild-type (WT) and p.Ile113del mutant TBX6 cDNAs were synthesized by BioVision (Shanghai, China) and cloned into the pcDNA3.1-Flag vector using the Hieff Clone Plus One Step Cloning Kit (Yeasen Biotechnology Co., Ltd., Shanghai, China), following the manufacturer’s instructions. The TBX6-p.Met111Ile mutant constructs was generated using a Mut Express II Fast Mutagenesis Kit V2 (Vazyme, Nan**g, Jiangsu, China). A 300-bp MESP2 promoter sequence was synthesized by BioVision and cloned into the pGL3-basic vector (Promega, Madison, WI, USA) to generate a luciferase reporter plasmid (MESP2-P2L-300-luc). Verification of all vectors was carried out through Sanger sequencing. Human embryonic kidney (HEK) 293T cells were obtained from the ATCC (American Type Culture Collection, Manassas, VA, USA). 293T cells were cultured in Dulbecco’s modified Eagle’s medium (Corning, NY, USA), supplemented with 10% (v/v) fetal bovine serum (BioVision Technology (Shanghai) Co., Ltd., Shanghai, China) and 1% penicillin/streptomycin (Beyotime Biotechnology, Shanghai, China) in a 5% CO2 incubator at 37 °C. Transfection was performed using jetPRIME transfection reagent (Polyplus Transfection, Illkirch, France). HEK 293T cells were seeded (5 × 105 cells/well) in 12-well plates and transfected with 0.8 µg of the designated TBX6 constructs. At 36 h post-transfection, proteins were extracted from whole-cell lysates, separated on 10% sodium dodecyl-sulfate polyacrylamide gel electrophoresis gels, transferred to polyvinylidene difluoride membranes, and incubated with rabbit anti-Flag polyclonal antibody (Proteintech, Wuhan, Hubei, China). Protein detection was achieved using a chemiluminescence system with a horseradish peroxidase-conjugated secondary antibody. β-actin was used as a control for ensuring consistent loading. The MESP2-P2L-300-luc plasmid was employed to assess the regulatory role of TBX6 in transcriptional modulation. Briefly, HEK293T cells were seeded in 96-well plates at a density of 4 × 104 per well. After 24 h of culture, the cells were co-transfected with 80 ng of MESP2-P2L-300-luc, 1 ng of pRL-SV40, and 20 ng of control or TBX6 expression vectors. Subsequently, 24 h after transfection, the luciferase activity was measured using the Dua-Glo luciferase assay system (Promega Corporation, Madison, WI, USA) and normalized to Renilla luciferase encoded by pRL-SV40.In silico analysis of the TBX6‑Ile113del variant
Plasmids construction
Cell culture and transient transfection
Western blot analysis
Luciferase assay
Results
Description of the fetus
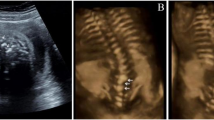
The fetus was the first pregnancy of the family, and there was no special in the prenatal examination before 12 weeks of gestation. At 13 weeks of gestation, the fetus was first shown to have abnormal spinal development by ultrasound examination at local hospital. A detailed fetal ultrasound scan at 17 weeks of gestation at our hospital revealed abnormal development of the spinal cord, unusual arrangement of the local vertebrae in the thoracic and sacrococcygeal segments, and scoliosis (Fig. 1). No abnormalities were found in other organs of the fetus. After receiving detailed genetic counseling about the results of trio-WES and CMA, the pregnant woman chose to terminate the pregnancy at 23 weeks. The autopsy was conducted and the results confirmed that the fetus had scoliosis (to the left), involving the thoracic_8 to lumbar_5 segments, accompanied by lumbar_5 vertebral hypertrophy.

Ultrasound scan of the fetus at 17 weeks of gestation
Identification and in silico analysis of the TBX6 variants in the patient
To explore the underlying genetic variants of the fetus, trio-WES was performed in the family. After excluding low-confidence variants, high-frequency variants with an allele frequency above 1% in the gnomAD database, and benign variants predicted by REVEL [12], a heterozygous in-frame deletion variant [NM_004608.4: c.338_340del, p.(Ile113del)] and a well-known homozygous hypomorphic allele (T-C-A) in TBX6 gene were identified in the fetal genome. Sanger sequencing revealed that the c.338_340del variant was inherited from the mother, and both parents are heterozygous carriers of the hypomorphic allele (Fig. 2). Analysis of the sequencing data showed that the two heterozygous variants are cis in the mother.

Pedigree and chromatogram of Sanger sequencing analysis of a Chinese family. (a) Family pedigree. (b-1) A heterozygous TBX6 variant (c.338_340del, p.Ile113del) in the fetus and the mother. (b-2) The homozygous common hypomorphic allele in the fetus, and the heterozygous common hypomorphic allele in the mother and father. Red arrows indicate the corresponding sequences where the mutation was found
The TBX6 p.Ile113del variant has not been reported previously and was not included in the known public databases (i.e., gnomAD, HGMD, and ClinVar). Ile113 resides within the T-box domain (DNA-binding region) of TBX6 and is highly conserved in multiple species (Fig. 3a). Predictive assessments using various in silico tools, including MutationTaster2 and MutPred-Indel, indicated the deleterious nature of the p.Ile113del variant. In addition, the crystal structure analysis using TBX3 homologous sequence showed that the p.Ile113del variant likely impaired its DNA-binding ability (Fig. 3b).

(a) Conservation analysis of TBX6. Alignment of amino acid sequences of TBX6 in different species. The circled residues represent the corresponding residues of TBX6 p.Ile113. (b)Space distribution of the two variants evaluated in this study. A T-box domain model of TBX6 (89–273 amino acids) in complex with DNA was built using the Swiss-Model with the crystal structure of TBX3 (PDB ID code: 1H6F) as a reference. The the two variants (p.Met111Ile and p.Ile113del) reside in the T-box domain of the TBX6 protein, and are in close proximity to the target DNA
Impairment of the transcriptional activity of TBX6 by the Ile113del variant
To assess the impact of the p.Ile113del variant on TBX6 function, a luciferase assay was performed using a reporter construct containing a fragment of the MESP2 promoter as described [3]. In addition to the TBX6 p.Ile113del variant, a previously reported deleterious missense variant (Met111Ile), located close to Ile113, was also constructed as a positive control [6]. In vitro experiments showed that these two variants did not impair the protein expression of TBX6, but rather upregulated the protein expression levels to varying degrees (Fig. 4a). As expected, the Met111Ile mutant displayed extremely low transcriptional activity, suggesting that the functional evaluation system was appropriate. Compared with WT-TBX6, the Ile113del variant almost resulted in complete loss of its transcriptional activity (Fig. 4b). Together, these results indicate that although the Ile113del mutant protein only loses one amino acid, its impact on TBX6 is similar to those of null variants (i.e., nonsense variants), greatly impairing its transcriptional activity. According to the guidelines recommended by the American College of Medical Genetics and Genomics (ACMG) for the interpretation of sequence variants [13], the variant (c.338_340del) was classified as likely pathogenic (PS3_Supporting + PM1 + PM2_Supporting + PM4 + PP4). This in-frame deletion variant, together with a known hypomorphic haplotype (T-C-A) in TBX6 could explain the CS in the fetus.

Results of the western blot and luciferase assay. (a) The expression of the WT and mutant Flag-TBX6 was evaluated using a western blot assay. (b) Luciferase transactivation assays of the WT and mutant Flag-TBX6 using MESP2 promoter reporter. Triple asterisks (***) indicate p values less than 0.0001
Identification of 17q12 microdeletion in the family as a secondary finding
It has been reported that approximately 5% of the patients have more than one molecular finding [14]. We conducted routine analysis of CNVs for each sample by comparing the read depth with WES data from other samples of the same batch. By this strategy, we identified a 1.85 Mb deletion on 17q12 region (hg19, chr17:34493374–36,346,847) in the fetus and the mother (Fig. 5a), which was also identified by CMA. This CNV can lead to 17q12 microdeletion syndrome, mainly manifested as renal cysts and diabetes syndrome caused by haploinsufficiency of HNF1B gene (chr17:34815072–36,192,489, OMIM#189,907) (Fig. 5b) [15]. According to the clinical interpretation guidelines of the American College of Medical Genetics and Genomics and the Clinical Genome [16], the microdeletion of 17q12 was classified as pathogenic.

Copy number variation analysis in this family. (a) Sequencing depth analysis using the WES data showed a 17q12 deletion (hg19, chr17:34493374–36,346,847) in the fetus and mother harbored. (b) Genes involved in the 17q12 region
Since the ultrasound examination did not indicate any renal abnormalities in the fetus, we therefore evaluated the health status of the mother. Ultrasound reports from another hospital showed the mother had multiple cystic areas in both kidneys with multiple stones, suggesting cystic changes in the renal medulla, the possibility of a sponge kidney, and several anechoic areas in the right kidney, indicative of multiple renal cysts.
Discussion
In this study, we reported a Chinese fetal patient with spinal abnormalities who was concurrent of compound heterozygous variants of a novel in-frame deletion (c.338_340del, p.Ile113del) and the common hypomorphic haplotype in TBX6 and a 1.85 Mb deletion on 17q12 region (hg19, chr17:34493374–36,346,847). Postnatal patients with TACS have been widely reported in Han Chinese, Japanese and European cohorts, and a clinical diagnostic algorithm (TACScore) had been developed [7]. The affected individuals can present with a range of spinal deformity from simple hemivertebrae/butterfly vertebrae involving the lower part of the spine (T8-S5), with or without simple rib anomaly to a more severe clinical phenotype, defined as spondylocostal dysostosis (SCD) that involves multiple segmentation defects of the vertebrae with malalignment of the ribs [7]. In contrast, few prenatal cases were reported. In 2020, Errichiello et al. reported a SCD fetus with severe kyphoscoliosis involving the entire spine and segmentation abnormalities (hemivertebrae) accompanied by asymmetric rib defects, who harbored a homozygous TBX6 stop-gained variant [NM_004608.3:c.1148 C > A, p.(Ser383Ter)] [17]. And in 2024, Liu et al. reported five fetuses with vertebral malformations (such as hemivertebra, butterfly vertebra, and scoliosis), who were detected with 16p11.2 deletion and the TBX6 haplotype of T-C-A. However, no detailed phenotypic description was available and the clinical information was limited [18]. The understanding of the prenatal phenotype of TACS is still limited and requires more collection of fetal patients.
In reported studies of TACS, the rare null mutations of TBX6 include 16p11.2 deletion, frameshift variants, nonsense variants, splicing variants, and missense variants. To the best of our knowledge, no deleterious single amino acid deletion variant has been reported in patients with TACS. The present study reported the first pathogenic single amino acid deletion variant of TBX6 (c.338_340del, p.Ile113del). Assessing the pathogenicity of single amino acid deletion variants, akin to missense variants, poses challenges not encountered with apparent null variants. Therefore, conducting functional research is important for evaluating the variants. Given its role as a transcription factor, the transcriptional assay was suitable for the assessment of functional change in TBX6. We here clearly showed the Ile113del variant led to the loss of TBX6 transcriptional activity, indicating its characterization as a null variant. Additionally, analysis of the 3D structure of TBX6 showed that the Ile113 and Met111 are very close and both have the ability to directly bind to DNA (Fig. 3), which implies that the functional changes of Ile113del may be in line with that of the deleterious variant in the 111 residue. The results from luciferase report assays have well confirmed this hypothesis (Fig. 4). Considering the time challenge of functional experiments in prenatal diagnosis, we emphasize deepening our understanding of variant pathogenicity through structural analysis.
TACS is caused by bi-allelic variants of TBX6, consistent with autosomal recessive trait manifestation. The only exception was an autosomal dominant SCD caused by TBX6 elongation (c.1311 A > T, p.*437Cext*81) variant reported in a Macedonian family, whose pathogenic mechanism remained to be explored. To explain the heterogeneity of the TACS clinical phenotype, an allelic heterogeneity hypothesis of TBX6 was proposed. For the heterozygous common risk haplotype of TBX6, it causes only a moderate reduction in TBX6 expression. At the same time, the TBX6 mutations in the other allele lead to different dosage effects, thus causing variation of phenotypic severity [19].
In addition to the compound heterozygosity in TBX6, the fetus harbored a microdeletion of 17q12 inherited from the mother. The 17q12 deletion syndrome can lead to a variety of clinical phenotypes after birth, mainly including congenital abnormalities of the kidney and urinary tract (genital tract abnormalities, renal cysts, abnormalities of kidney parenchyma, fusion anomalies), MODY5, and neurodevelopmental or neuropsychiatric disorders (developmental delay, intellectual disability, autism spectrum disorder, schizophrenia) [10]. However, the knowledge about the prenatal phenotypes of 17q12 deletion syndrome is still limited. The results of ultrasound screening at gestational weeks of 20–32 showed that the most common prenatal phenotypes were kidney anomalies, including renal hyperechogenicity and multicystic dysplastic kidneys [20, 21]. Although ultrasound scan of the fetus in this study at 17 weeks of gestation found no abnormalities on kidneys, it was possible to appear kidney anomalies after 20 weeks. Moreover, it was reported that the penetrance of 17q12 deletion was 34.4% (13.7-70.0%) [22], which posed challenges for prenatal genetic counseling. The 17q12 deletion syndrome is not involved in congenital scoliosis. Therefore, the CNV of the 17q12 deletion was identified as the cause of the mother’s manifestation of multiple renal cysts, with no discernable connection with the congenital scoliosis of the fetus.
In conclusion, our study identified a novel TBX6 variant with a common risk haplotype and elucidated the damaging effect of the indel variant on the TBX6 protein via in vitro experiments. This study is the first to report a Chinese patient with TACS during the fetal period, which broadens the mutation spectrum of the TBX6 gene and extends the genotype-phenotype relationship in the study of TACS. In addition, we yet reported a pathogenic CNV with clinical penetrance in the fetus, indicating once again the complexity of prenatal diagnosis and genetic counseling.
Data availability
The raw data are available from the corresponding author on reasonable request.
Abbreviations
- TBX6:
-
Transcription Factor Box 6
- CS:
-
Congenital Scoliosis
- SCD:
-
Spondylocostal Dysostosis
- TACS:
-
TBX6-Associated Congenital Scoliosis
- CMA:
-
Microarray Analysis
- WES:
-
Whole-Exome Sequencing
- CNV:
-
Copy Number Variation
- SNPs:
-
Single Nucleotide Polymorphisms
- 3D:
-
Three-Dimensional
- WT:
-
Wild-Type
- HEK:
-
Human Embryonic Kidney
- MODY5:
-
Maturity-Onset Diabetes Of The Young Type 5
References
Hedequist D, Emans J, J Am Acad Orthop Surg. Congenital scoliosis. 2004 Jul-Aug;12(4):266 – 75. https://doi.org/10.5435/00124635-200407000-00007. PMID: 15473678.
Giampietro PF, Dunwoodie SL, Kusumi K, Pourquié O, Tassy O, Offiah AC, Cornier AS, Alman BA, Blank RD, Raggio CL, Glurich I, Turnpenny PD. Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans. Ann N Y Acad Sci. 2009;1151:38–67. https://doi.org/10.1111/j.1749-6632.2008.03452.x. PMID: 19154516.
Turnpenny PD, Alman B, Cornier AS, Giampietro PF, Offiah A, Tassy O, Pourquié O, Kusumi K, Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev Dyn. 2007;236(6):1456-74. https://doi.org/10.1002/dvdy.21182. PMID: 17497699.
Wu N, Ming X, **ao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, **e H, Gucev ZS, Liu S, Yang N, Al-Kateb H, Chen J, Zhang J, Hauser N, Zhang T, Tasic V, Liu P, Su X, Pan X, Liu C, Wang L, Shen J, Shen J, Chen Y, Zhang T, Zhang J, Choy KW, Wang J, Wang Q, Li S, Zhou W, Guo J, Wang Y, Zhang C, Zhao H, An Y, Zhao Y, Wang J, Liu Z, Zuo Y, Tian Y, Weng X, Sutton VR, Wang H, Ming Y, Kulkarni S, Zhong TP, Giampietro PF, Dunwoodie SL, Cheung SW, Zhang X, ** L, Lupski JR, Qiu G, Zhang F. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015;372(4):341–50. https://doi.org/10.1056/NEJMoa1406829. Epub 2015 Jan 7. PMID: 25564734; PMCID: PMC4326244.
Lefebvre M, Duffourd Y, Jouan T, Poe C, Jean-Marçais N, Verloes A, St-Onge J, Riviere JB, Petit F, Pierquin G, Demeer B, Callier P, Thauvin-Robinet C, Faivre L, Thevenon J. Autosomal recessive variations of TBX6, from congenital scoliosis to spondylocostal dysostosis. Clin Genet. 2017;91(6):908–12. https://doi.org/10.1111/cge.12918. Epub 2017 Feb 22. PMID: 27861764.
Takeda K, Kou I, Kawakami N, Iida A, Nakajima M, Ogura Y, Imagawa E, Miyake N, Matsumoto N, Yasuhiko Y, Sudo H, Kotani T, Japan Early Onset Scoliosis Research Group, Nakamura M, Matsumoto M, Watanabe K, Ikegawa S. Compound heterozygosity for null mutations and a common hypomorphic risk haplotype in TBX6 causes congenital scoliosis. Hum Mutat. 2017;38(3):317–23. https://doi.org/10.1002/humu.23168. Epub 2017 Jan 18. PMID: 28054739.
Liu J, Wu N, Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) study, Yang N, Takeda K, Chen W, Li W, Du R, Liu S, Zhou Y, Zhang L, Liu Z, Zuo Y, Zhao S, Blank R, Pehlivan D, Dong S, Zhang J, Shen J, Si N, Wang Y, Liu G, Li S, Zhao Y, Zhao H, Chen Y, Zhao Y, Song X, Hu J, Lin M, Tian Y, Yuan B, Yu K, Niu Y, Yu B, Li X, Chen J, Yan Z, Zhu Q, Meng X, Chen X, Su J, Zhao X, Wang X, Ming Y, Li X, Raggio CL, Zhang B, Weng X, Zhang S, Zhang X, Watanabe K, Matsumoto M, ** L, Shen Y, Sobreira NL, Posey JE, Giampietro PF, Valle D, Liu P, Wu Z, Ikegawa S, Lupski JR, Zhang F, Qiu G. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet Med. 2019;21(7):1548–58. https://doi.org/10.1038/s41436-018-0377-x. Epub 2019 Jan 14. PMID: 30636772; PMCID: PMC6659397.
Yang N, Wu N, Zhang L, Zhao Y, Liu J, Liang X, Ren X, Li W, Chen W, Dong S, Zhao S, Lin J, **ang H, Xue H, Chen L, Sun H, Zhang J, Shi J, Zhang S, Lu D, Wu X, ** L, Ding J, Qiu G, Wu Z, Lupski JR, Zhang F. TBX6 compound inheritance leads to congenital vertebral malformations in humans and mice. Hum Mol Genet. 2019;28(4):539–47. https://doi.org/10.1093/hmg/ddy358. PMID: 30307510; PMCID: PMC6489408.
Yasuhiko Y, Haraguchi S, Kitajima S, Takahashi Y, Kanno J, Saga Y. Tbx6-mediated notch signaling controls somite-specific Mesp2 expression. Proc Natl Acad Sci U S A. 2006;103(10):3651–6. https://doi.org/10.1073/pnas.0508238103. Epub 2006 Feb 27. PMID: 16505380; PMCID: PMC1450137.
Mitchel MW, Moreno-De-Luca D, Myers SM et al. 17q12 Recurrent Deletion Syndrome. 2016 Dec 8 [Updated 2020 Oct 15]. In: Adam MP, Feldman J, Mirzaa GM, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024.
Li S, Han X, Wang Y, Chen S, Niu J, Qian Z, Li P, ** L, Xu C. Chromosomal microarray analysis in fetuses with congenital anomalies of the kidney and urinary tract: a prospective cohort study and meta-analysis. Prenat Diagn. 2019;39(3):165–74. Epub 2019 Feb 7. PMID: 30650192.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W. REVEL: an Ensemble Method for Predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–85. Epub 2016 Sep 22. PMID: 27666373; PMCID: PMC5065685.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, **ao R, Ding Y, **a F, Beaudet AL, Muzny DM, Gibbs RA, Boerwinkle E, Eng CM, Sutton VR, Shaw CA, Plon SE, Yang Y, Lupski JR. Resolution of Disease Phenotypes resulting from Multilocus genomic variation. N Engl J Med. 2017;376(1):21–31. https://doi.org/10.1056/NEJMoa1516767. Epub 2016 Dec 7. PMID: 27959697; PMCID: PMC5335876.
Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, Woolf AS, Rizzoni G, Novelli G, Nicholls AJ, Hattersley AT. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68(1):219–24. Epub 2000 Nov 20. PMID: 11085914; PMCID: PMC1234916.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–257. https://doi.org/10.1038/s41436-019-0686-8. Epub 2019 Nov 6. Erratum in: Genet Med. 2021;23(11):2230. PMID: 31690835; PMCID: PMC7313390.
Errichiello E, Arossa A, Iasci A, Villa R, Ischia B, Pavesi MA, Rizzuti T, Bedeschi MF, Zuffardi O. An additional piece in the TBX6 gene dosage model: a novel nonsense variant in a fetus with severe spondylocostal dysostosis. Clin Genet. 2020;98(6):628–9. https://doi.org/10.1111/cge.13854. Epub 2020 Oct 14. PMID: 33058178.
Liu L, Wang J, Liu X, Wang J, Chen L, Zhu H, Mai J, Hu T, Liu S. Prenatal prevalence and postnatal manifestations of 16p11.2 deletions: a new insights into neurodevelopmental disorders based on clinical investigations combined with multi-omics analysis. Clin Chim Acta. 2024;552:117671. https://doi.org/10.1016/j.cca.2023.117671. Epub 2023 Nov 19. PMID: 37984529.
Chen W, Lin J, Wang L, Li X, Zhao S, Liu J, Akdemir ZC, Zhao Y, Du R, Ye Y, Song X, Zhang Y, Yan Z, Yang X, Lin M, Shen J, Wang S, Gao N, Yang Y, Liu Y, Li W, Liu J, Zhang N, Yang X, Xu Y, Zhang J, Delgado MR, Posey JE, Qiu G, Rios JJ, Liu P, Wise CA, Zhang F, Wu Z, Lupski JR, Wu N. TBX6 missense variants expand the mutational spectrum in a non-mendelian inheritance disease. Hum Mutat. 2020;41(1):182–95. https://doi.org/10.1002/humu.23907. Epub 2019 Sep 26. PMID: 31471994; PMCID: PMC7061259.
Zhou CX, Zhu XY, Zhu YJ, Gu LL, He LL, Liu W, Yang Y, Wu X, Duan HL, Ru T, Li J. Prenatal features of 17q12 microdeletion and microduplication syndromes: A retrospective case series. Taiwan J Obstet Gynecol. 2021;60(2):232–237. https://doi.org/10.1016/j.tjog.2021.01.001. PMID: 33678321.
**g XY, Huang LY, Zhen L, Han J, Li DZ. Prenatal diagnosis of 17q12 deletion syndrome: a retrospective case series. J Obstet Gynaecol. 2019;39(3):323–327. doi: 10.1080/01443615.2018.1519693. Epub 2019 Jan 11. PMID: 30634886.
Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. 2013;15(6):478–81. https://doi.org/10.1038/gim.2012.164. Epub 2012 Dec 20. PMID: 23258348; PMCID: PMC3664238.
Funding
This study was funded by Shanghai Science and Technology Innovation Action Plan (22Y11906700).
Author information
Authors and Affiliations
Contributions
Shuyuan Li and Niu Li designed the study. Guoqiang Li, Yiyao Chen and Xu Han performed experiments. Guoqiang Li wrote the first draft of the manuscript. Shuyuan Li and Niu Li revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of International Peace Maternity and Child Health Hospital, and this study was performed in accordance with relevant guidelines. Informed consent was obtained from the parents of the fetus.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, G., Chen, Y., Han, X. et al. Concurrent of compound heterozygous variant of a novel in-frame deletion and the common hypomorphic haplotype in TBX6 and inherited 17q12 microdeletion in a fetus. BMC Pregnancy Childbirth 24, 456 (2024). https://doi.org/10.1186/s12884-024-06653-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-024-06653-2