
Abstract
Background
Acquiring comprehensive insight into the dynamics of Mycobacterium tuberculosis (Mtb) population structure is an essential step to adopt effective tuberculosis (TB) control strategies and improve therapeutic methods and vaccines. Accordingly, we performed this systematic review and meta-analysis to determine the overall prevalence of Mtb genotypes/ sublineages in Iran.
Methods
We carried out a comprehensive literature search using the international databases of MEDLINE and Scopus as well as Iranian databases. Articles published until April 2020 were selected based on the PRISMA flow diagram. The overall prevalence of the Mtb genotypes/sublineage in Iran was determined using the random effects or fixed effect model. The metafor R package and MedCalc software were employed for performing this meta-analysis.
Results
We identified 34 studies for inclusion in this study, containing 8329 clinical samples. Based on the pooled prevalence of the Mtb genotypes, NEW1 (21.94, 95% CI: 16.41–28.05%), CAS (19.21, 95% CI: 14.95–23.86%), EAI (12.95, 95% CI: 7.58–19.47%), and T (12.16, 95% CI: 9.18–15.50%) were characterized as the dominant circulating genotypes in Iran. West African (L 5/6), Cameroon, TUR and H37Rv were identified as genotypes with the lowest prevalence in Iran (< 2%). The highest pooled prevalence rates of multidrug-resistant strains were related to Bei**g (2.52, 95% CI) and CAS (1.21, 95% CI).
Conclusions
This systematic review showed that Mtb populations are genetically diverse in Iran, and further studies are needed to gain a better insight into the national diversity of Mtb populations and their drug resistance pattern.
Similar content being viewed by others


Background
Tuberculosis (TB) remains the most lethal infectious disease with an estimated rate of 1.4 million deaths in 2018 [1]. Human-adapted Mycobacterium tuberculosis complex (MTBC), as a causative agent of TB infection, belong to eight phylogenetic branches with a phylogeographical population structure [2, 3]. These lineages include Indo-Oceanic lineage (Lineage 1), East Asian (Lineage 2), Central Asian (Lineage 3), Euro-American (Lineage 4), Ethiopian (Lineage 7), known as Mycobacterium tuberculosis sensu stricto, West African 1 (Lineage 5) and West African 2 (Lineage 6), referred to as Mycobacterium africanum and Lineage 8 (L8) which geographically restricted to the African Great Lakes region [2,3,4].
Different studies have shown that genomic differences among MTBC lineages or sublineages can affect the clinical and epidemiological characteristics of TB infection [5,6,7,8]. In recent decades, some Mycobacterium tuberculosis (Mtb) lineages/sublineages have attracted wide attention due to certain features such as transmission potential, pathogenic properties and association with drug resistance [9, 10]. Lineages 2 and 4 are widely distributed and seem to have a higher pathogenic power compared to geographically restricted lineages [2, 11, 12]. In West and South Asia, a sharp increase has been documented in the circulation of certain sublinages such as NEW-1 (Lineage 4) and CAS (Lineage 3) strains that are prone to emerging as resistant clones [13,14,15]. This growing increase seems be more important in Iran with the national average TB rate of 14 cases per 100,000 population, due to the influx of Afghan refugees and population growth [1]. Accordingly, acquiring comprehensive insight into the dynamics of Mtb population structure is an essential step to adopt effective TB control strategies and improve therapeutic methods and vaccines.
Therefore, the current systematic review and meta-analysis was conducted to determine (1) the overall prevalence of Mtb genotypes/sublineages and (2) to determine the dominant multidrug-resistant (MDR) Mtb genotypes in TB patients in Iran.
Methods
Study protocol
The meta-analysis was based on the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines for systematic reviews and meta-analyses [16]. The study protocol was registered in the PROSPERO database (CRD42020186561).
Search strategy and selection criteria
For evaluating the diversity of Mtb isolates in Iran, a comprehensive literature search was conducted using the international electronic databases of MEDLINE and Scopus as well as Iranian databases. English-language studies published until April 2020 were retrieved using the following keywords: “Mycobacterium tuberculosis”, “tuberculosis”, “molecular ty**”, “genetic diversity”, “genoty**” and “Iran” combined with the Boolean operators “OR”, “AND” and “NOT” in the Title/Abstract/Keywords field. Additional keywords such as “lineage” combined with “Mycobacterium tuberculosis” were used to avoid missing any articles. Similar strategies using Persian keywords were used to find relevant Persian original articles in Iranian databases, such as Scientific Information Database (www.sid.ir), Irandoc (www.irandoc.ac.ir), Magiran (www.magiran.com), and Iranmedex (www.iranmedex.com).
The titles and abstracts of all the identified articles were reviewed for eligibility, then screening for relevant articles were performed by reviewing the full texts.
The inclusion criteria were: 1) studies reporting the prevalence of Mtb genotypes among TB patients, 2) studies presenting data from Iran irrespective of the publication year, and 3) studies used Spoligoty**, mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) ty** and Whole-Genome Sequencing (WGS) methods for genoty**. The exclusion criteria, on the other hand, included:1) studies only presenting prevalence data on Mtb genotypes among drug-resistant Mtb isolates, 2) studies providing incomplete data, 3) studies published as meta-analyses and systematic reviews, 4) studies not in English or Persian, 5) studies limited to a single genotype, 6) studies that lacked genoty** data, and 7) studies that were not related to human TB molecular epidemiology. Data screening was performed by two reviewers independently.
Data extraction and quality assessment
Data from the studies meeting our inclusion criteria were extracted. We required the following data: first author’s name, year of publication, study area, molecular techniques, genotype, number of genotypes, total sample size, MDR genotype, sample type and nationality.
According to the items defined in the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist, we evaluated the methodological quality of the included studies using the pre-defined criteria presented in Table 1. This checklist consists of various methodological aspects, and a maximum quality evaluation score of 32 was considered and articles with scores below 18 were excluded from this study [51]. Data extraction and quality assessment were also carried out by two reviewers independently.
Statistical analysis
Pooled proportion and 95% CI were used to assess the prevalence of the genotypes in the pulmonary tuberculosis (PTB) and extrapulmonary tuberculosis (EPTB) samples. Generalized linear mixed model with random intercept logistic regression model was used for assessing pooled prevalence [52]. The heterogeneity of prevalence between the included studies was tested and quantified by using Cochran’s Q test and I2 index, respectively [53]. Clopper-Pearson was run for evaluating pooled proportion and confidence interval in the individual studies. Also, continuity correction of 0.5 was considered in studies with zero cell frequencies [54]. The pooled proportion, as an overall prevalence of the genotypes, was derived by the random effects model because of significant heterogeneity between the individual studies. Publication bias was tested by Egger’s linear regression test and Begg’s test (P < 0.05 was set as the significance level for publication bias) [55]. All the statistical analyses were performed by using the metafor R package and MedCalc software.
Results
Search results and studies’ characteristics
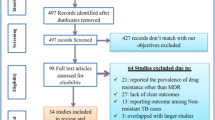
A total of 316 articles were identified by the primary search strategy, of which 34 articles met the eligibility criteria and were included in this study (Fig. 1). The selected studies included 8329 clinical samples. Most of the studies were conducted in Tehran (capital of Iran). Publication year of these studies ranged from 2006 to 2020. Spoligoty** and MIRU-VNTR ty** were identified as the most common methods of genoty**.

Flow diagram of literature selection process in the meta-analysis
Quality assessment
Based on the scores of the STROBE checklist, the highest and lowest scores were related to the studies of Velayati et al. (2014) and Feyisa et al. (2016), respectively. The mean score of STROBE tool was 25.72 (SD = 2.42, range: 20–31) (Table 1).
Pooled prevalence of MTBC genotypes in the PTB and EPTB samples
Results of the random or fixed effects meta-analysis are summarized in Table 2. M. bovis as a member of the animal-adapted MTBC accounted for only 3.29% of the studied strains and Mtb sensu stricto (Lineages1–4) comprised the largest proportion of the studied strains. Based on the pooled prevalence of the Mtb genotypes in the PTB and EPTB samples, NEW1 (21.94, 95% CI: 16.41–28.05%), CAS (19.21, 95% CI: 14.95–23.86%), EAI (12.95, 95% CI: 7.58–19.47%), and T (12.16, 95% CI: 9.18–15.50%) were found to be the dominant circulating genotypes in Iran. West African (L 5/6), Cameroon, TUR and H37Rv (parts of the Euro-American super-lineage [L4]) were identified as genotypes with the lowest prevalence in Iran (< 2%). The forest plot of some of the genotypes (i.e., Bei**g, CAS, and EAI) are shown in Fig. 2. In addition, the highest pooled prevalence of MDR strains was found in Bei**g (2.52,95% CI) and CAS (1.21,95% CI) genotypes (Table 2).

Forest plots displaying the prevalence of different M.tb genotypes in the studied geographical region
Publication bias
We observed significant heterogeneity across the studies based on the I2 index with a few exceptions (Table 2). However, publication bias was not significant based on the results of Egger’s linear regression test and Begg’s test.
Discussion
Based on the pooled data investigated, all MTBC lineages, except lineage 7 and 8, were found in Iran, which reflects the presence of high diversity in MTBC strains. Phylogeographical population structure of the MTBC stems from the interplay between different factors such as human migration, geography, genetic drift and host-pathogen interaction [4, 5, 56]. Iran is the main host country for Afghan refugees, but the main factor contributing to formation of MTBC lineages phylogeography in Iran has not been identified.
Study of global variation in MTBC strains showed that the prevalence of lineages 2, 3 and 4 strains may be increasing in West Asia, while the prevalence of lineage 1 is declining [15]. The summary of Mtb strains diversity in Iran, based on families/sublineage, showed that NEW1(L4) (21.94, 95% CI: 16.41–28.05%), CAS (L3) (19.21, 95% CI: 14.95–23.86%), EAI(L1) (12.95, 95% CI: 7.58–19.47%), and T (L4) (12.16, 95% CI: 9.18–15.50%) were the dominant circulating Mtb genotypes. EAI (L1) and CAS (L3) are mainly confined to the areas around the Indian Ocean [11]. Movement of strains with people from these regions may explain the presence of these genotypes in Iran. Besides, appearing CAS as a one of the prevalent Mtb subpopulations in Iran may reflect the pathogenic properties of this genotype.
In a recent study, the global proportion of MDR in CAS population was estimated at 30.63% [57]. In our study, based on the pooled prevalence of MDR genotype, CAS was found (1.21%) as a one of the dominant genotypes. This finding reflects the needs for more understanding and monitoring of this subpopulation.
Despite the global dissemination of Bei**g genotype as a prototype of lineage 2, it had low prevalence in our geographical region. However, the highest pooled prevalence of MDR strains was found in the Bei**g (2.52%) genotype. This result is consistent with the previously published reports about the prevalence of Bei**g among MDR-TB isolates in Iran [58]. The low prevalence of Bei**g genotype compared to other genotypes in Iran may be explained by the prevalent Bei**g sublineage, affecting its pathobiological properties and epidemiological dynamics. Further studies are warranted to identify the distribution pattern of the Bei**g sublineages in Iran, which can improve the management of their infection.
The dominance of NEW1 as a specialist sublineage of Euro-American lineage (L4) in Iran was not unexpected. Some evidence has shown that Iran is the probable origin of this family/sublineage, which may reflect ecological adaption in this subpopulation [59]. It is noteworthy that NEW1 genotype is prone to MDR [13]. The pooled prevalence of MDR in NEW1 was 0.8% (95% CI). However, the results of overall MDR estimation may be less representative of the target population, as in the some of the included studies in our analysis; drug susceptibility testing was not reported based on the identified genotype, which may lead to variation in the final results. Other sublineages of lineage 4 such as T, Haarlem, Uganda and S in varying proportions were also observed. This distribution pattern in the subtypes of lineage 4 in Iran may be explained by the effect of human migration and genetic and phenotypic characteristics of each sublineage.
In addition, we observed that lineage 5/6 subtype had the lowest prevalence in our geographical region. Based on the fact that these strains are geographically restricted [2], we can only speculate human migration as the determinant of this distribution. A limitation of this study is that most of the included studies were conducted in Tehran (Capital of Iran). Thus, our finding may not be completely representative of the overall prevalence of different Mtb populations in Iran. In addition, the most of the included studies were based on Spoligoty** and MIRU-VNTR ty** methods while WGS provides a superior resolution compared with these PCR-based genoty** methods to identification of diversity in Mtb strains.
Conclusions
In summary, this systematic review showed that Mtb population are genetically diverse in Iran and the NEW1 (L4) and West African (L5/6) genotypes had the highest and lowest pooled prevalence rates, respectively. This type of evidence can contribute to better clinical and epidemiological management of Mtb infections. Also, there is a need for further in-depth studies to gain a deeper insight into the national diversity of Mtb populations and their drug resistance pattern.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- MTBC:
-
Mycobacterium tuberculosis complex
- Mtb :
-
Mycobacterium tuberculosis
- MIRU-VNTR:
-
mycobacterial interspersed repetitive unit-variable number tandem repeat
- TB:
-
Tuberculosis
- MDR:
-
multidrug-resistant
- PRISMA:
-
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- STROBE:
-
Strengthening the Reporting of Observational Studies in Epidemiology
- PTB:
-
Pulmonary tuberculosis
- EPTB:
-
Extrapulmonary tuberculosis
- WGS:
-
Whole-Genome Sequencing
References
WHO. WHO global report, global tuberculosis report 2019. Geneva: World Health Organization; 2019.
Brites D, Gagneux S. The nature and evolution of genomic diversity in the mycobacterium tuberculosis complex. Adv Exp Med Biol. 2017;1019:1–26.
Ngabonziza JCS, Loiseau C, Marceau M, Jouet A, Menardo F, Tzfadia O, et al. A sister lineage of the mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat Commun. 2020;11(1):1–11.
Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, et al. Out-of-Africa migration and Neolithic coexpansion of mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45(10):1176.
Gagneux S, DeRiemer K, Van T, Kato-Maeda M, De Jong BC, Narayanan S, et al. Variable host–pathogen compatibility in mycobacterium tuberculosis. Proc Natl Acad Sci. 2006;103(8):2869–73.
Hadifar S, Behrouzi A, Fateh A, Khatami S, Rahimi Jamnani F, Siadat SD, et al. Comparative study of interruption of signaling pathways in lung epithelial cell by two different mycobacterium tuberculosis lineages. J Cell Physiol. 2019;234(4):4739–53.
Chapman HJ, Phillips SA, Hosford JL, Séraphin MN, Lauzardo M. Is the Bei**g strain of mycobacterium tuberculosis associated with cavitary lung disease? Infect Genet Evol. 2015;33:1–5.
Sarkar R, Lenders L, Wilkinson KA, Wilkinson RJ, Nicol MP. Modern lineages of mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PLOS ONE. 2012;7(8):e43170.
Hanekom M, Van Pittius NG, McEvoy C, Victor T, Van Helden P, Warren R. Mycobacterium tuberculosis Bei**g genotype: a template for success. Tuberculosis. 2011;91(6):510–23.
Brynildsrud OB, Pepperell CS, Suffys P, Grandjean L, Monteserin J, Debech N, et al. Global expansion of mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci Adv. 2018;4(10):eaat5869.
Coscolla M. Biological and epidemiological consequences of MTBC diversity. In: Gagneux S, editor. Strain variation in the Mycobacterium tuberculosis complex: its role in biology, epidemiology and control. Cham: Springer International Publishing; 2017. p. 95–116.
Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 2016;48(12):1535–43.
Mokrousov I. Emerging resistant clone of mycobacterium tuberculosis in West Asia. Lancet Infect Dis. 2016;16(12):1326–7.
Yasmin M, Gomgnimbou MK, Siddiqui RT, Refrégier G, Sola C. Multi-drug resistant mycobacterium tuberculosis complex genetic diversity and clues on recent transmission in Punjab, Pakistan. Infect Genet Evol. 2014;27:6–14.
Wiens KE, Woyczynski LP, Ledesma JR, Ross JM, Zenteno-Cuevas R, Goodridge A, et al. Global variation in bacterial strains that cause tuberculosis disease: a systematic review and meta-analysis. BMC Med. 2018;16(1):1–13.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264–9.
Amirmozafari N, Ramezanzadeh R, Farnia P, Ghazi F. The frequency of Bei**g genotype of mycobacterium tuberculosis isolated from tuberculosis patients. Razi J Med Sci. 2006;13(52):7–17.
Ramazanzadeh R, Amirmozafari N, Farnia P, Ghazi F. Genoty** of mycobacterium tuberculosis isolates from TB patients with spoligoty**. Scie J Kurdistan Univ Med Sci. 2006;11(1):50–9.
Masjedi MR, Varahram M, Mirsaeidi M, Ahmadi M, Khazampour M, Tabarsi P, et al. The recent-transmission of mycobacterium tuberculosis strains among Iranian and afghan relapse cases: a DNA-fingerprinting using RFLP and spoligoty**. BMC Infect Dis. 2008;8(1):109.
Tajeddin E, Farnia P, Kargar M, Noroozi J, Ahmadi M, Kazempour M, et al. Identification of mycobacterium tuberculosis Bei**g genotype using three different molecular methods. Koomesh. 2009;11(1):7–14.
Ramazanzadeh R, Farnia P, Amirmozafari N. Characterization of mycobacterium tuberculosis complex isolated from iranian and afghani patients by spoligoty** method. Braz J Microbiol. 2009;40(2):314–20.
Rohani M, Farnia P, Nasab MN, Moniri R, Torfeh M, Amiri M. Bei**g genotype and other predominant mycobacterium tuberculosis spoligotypes observed in Mashhad city, Iran. Indian J Med Microbiol. 2009;27(4):306.
Ahmadi M, Farnia P, Tajedin E, Tabarsi P, Baghaei P, Masjedi M, et al. Mycobacterium tuberculosis complex strains identification with Spoligoty** method in patients attending to Masih Daneshvari hospital. J Adv Med Biomed Res. 2009;17(67):23–32.
Jafarian M, Aghali-Merza M, Farnia P, Ahmadi M, Masjedi MR, Velayati AA. Synchronous comparison of mycobacterium tuberculosis epidemiology strains by “MIRU-VNTR” and “MIRU-VNTR and Spoligoty**” technique. Avicenna J Med Biotechnol. 2010;2(3):145.
Merza MA, Farnia P, Salih AM, Masjedi MR, Velayati AA. The most predominant spoligopatterns of mycobacterium tuberculosis isolates among Iranian, afghan-immigrant, Pakistani and Turkish tuberculosis patients: a comparative analysis. Chemotherapy. 2010;56(3):248–57.
Asgharzadeh M, Kafil HS, Roudsary AA, Hanifi GR. Tuberculosis transmission in northwest of Iran: using MIRU-VNTR, ETR-VNTR and IS6110-RFLP methods. Infect Genet Evol. 2011;11(1):124–31.
Jafarian M, Farnia P, Mozafari M, Ahmadi M, Masjiedi M, Velayati A. Comparison of the genetic convergence degree of mycobacterium tuberculosis (TB) strains isolated from patients infected with TB by MIRU-VNTR technique. J Adv Med Biomed Res. 2011;19(75):25–36.
Zaker Bostanabad S, Jabbarzadeh E, Pourazar S, Ghalami M. Ty** of mycobacterium tuberculosis isolated from patients in the Tehran City by Spoilgoty**. New Cellular Mol Biotechnol J. 2011;1(2):55–60.
Mozafari M, Farnia P, Jafarian M, Razavi Deligani M, Kazempour M, Masjedi M, et al. Comparison of mycobacterium tuberculosis Bei**g genotype with other mycobacterium tuberculosis strains using MIRU-VNTR method. ISMJ. 2012;15(1):1–12.
Haeili M, Darban-Sarokhalil D, Fooladi AAI, Javadpour S, Hashemi A, Siavoshi F, et al. Spoligoty** and drug resistance patterns of mycobacterium tuberculosis isolates from five provinces of Iran. Microbiologyopen. 2013;2(6):988–96.
Torkaman MRA, Nasiri MJ, FaRnia P, Shahhosseiny MH, Mozafari M, Velayati AA. Estimation of recent transmission of mycobacterium tuberculosis strains among Iranian and afghan immigrants: a cluster-based study. J Clin Diagn Res. 2014;8(9):DC05.
Velayati AA, Farnia P, Mozafari M, Sheikholeslami MF, Karahrudi MA, Tabarsi P, et al. High prevelance of rifampin-monoresistant tuberculosis: a retrospective analysis among Iranian pulmonary tuberculosis patients. Am J Trop Med Hyg. 2014;90(1):99–105.
Varahram M, Farnia P, Nasiri MJ, Karahrudi MA, Dizagie MK, Velayati AA. Association of Mycobacterium tuberculosis lineages with IFN-γ and TNF-α gene polymorphisms among pulmonary tuberculosis patient. Mediterranean J Hematol Infect Dis. 2014;6(1):e2014015–e.
Sharifipour E, Nasiri M, Farnia P, Mozafari M, Irani S. Evaluation of molecular diversity of mycobacterium tuberculosis strains by polymorphisms in RD regions. J Mycobac Dis. 2014;4(153):2161–1068.
Haeili M, Darban-Sarokhalil D, Fooladi AI, Zamani S, Zahednamazi F, Kardan J, et al. Genoty** and drug susceptibility testing of mycobacterium tuberculosis isolates from Iran. Int J Mycobacteriol. 2015;4:122.
Sharifipour E, Farnia P, Mozafari M, Irani S, Velayati AA. Deletion of region of difference 181 in mycobacterium tuberculosis Bei**g strains. Int J Mycobacteriol. 2016;5:S238–9.
Feyisa SG, Haeili M, Zahednamazi F, Mosavari N, Taheri MM, Hamzehloo G, et al. Molecular characterization of mycobacterium tuberculosis isolates from Tehran, Iran by restriction fragment length polymorphism analysis and spoligoty**. Rev Soc Bras Med Trop. 2016;49(2):204–10.
Zamani S, Haeili M, Nasiri MJ, Imani Fooladi AA, Javadpour S, Feizabadi MM. Genoty** of mycobacterium tuberculosis isolates from Hormozgan Province of Iran based on 15-locus MIRU-VNTR and Spoligoty**. Int J Bacteriol. 2016;2016:7146470.
Zaniani FR, Moghim S, Mirhendi H, Safaei HG, Fazeli H, Salehi M, et al. Genetic lineages of mycobacterium tuberculosis isolates in Isfahan, Iran. Curr Microbiol. 2017;74(1):14–21.
Ravansalar H, Tadayon K, Mosavari N, Derakhshan M, Ghazvini K. Genetic diversity of mycobacterium tuberculosis complex isolated from patients in the northeast of Iran by MIRU-VNTR and spoligoty**. Jundishapur J Microbiol. 2017;10(4):e39568.
Mansoori N, Yaseri M, Vaziri F, Douraghi M. Genetic diversity of mycobacterium tuberculosis complex isolates circulating in an area with high tuberculosis incidence: using 24-locus MIRU-VNTR method. Tuberculosis. 2018;112:89–97.
Azimi T, Nasiri MJ, Zamani S, Hashemi A, Goudarzi H, Fooladi AAI, et al. High genetic diversity among mycobacterium tuberculosis strains in Tehran, Iran. J Clin Tubercul Mycobacterial Dis. 2018;11:1–6.
Kamakoli MK, Khanipour S, Hadifar S, Ghajavand H, Farmanfarmaei G, Fateh A, et al. Challenge in direct Spoligoty** of mycobacterium tuberculosis: a problematic issue in the region with high prevalence of polyclonal infections. BMC Res Notes. 2018;11(1):486.
Babai Kochkaksaraei M, Kaboosi H, Ghaemi EA. Genetic variation of the mycobacterium tuberculosis in north of Iran; the Golestan Province. Iran Red Crescent Med J. 2019;21(8):e91553.
Hadifar S, Shamkhali L, Kamakoli MK, Mostafaei S, Khanipour S, Mansoori N, et al. Genetic diversity of mycobacterium tuberculosis isolates causing pulmonary and extrapulmonary tuberculosis in the capital of Iran. Mol Phylogenet Evol. 2019;132:46–52.
Afaghi GA, Moaddab SR, Darbouy M, Ansarian K, Hanifian S. Genotypic diversity of resistant mycobacterium tuberculosis strains isolated from tuberculosis patients in East Azerbaijan center by MIRU-VNTR. J Microb World. 2019;11(4):320–31.
Hadifar S, Kamakoli MK, Fateh A, Siadat SD, Vaziri F. Enhancing the differentiation of specific genotypes in mycobacterium tuberculosis population. Sci Rep. 2019;9(1):1–9.
Vaziri F, Kohl TA, Ghajavand H, Kargarpour Kamakoli M, Merker M, Hadifar S, et al. Genetic diversity of multi- and extensively drug-resistant mycobacterium tuberculosis isolates in the Capital of Iran, revealed by whole-genome sequencing. J Clin Microbiol. 2019;57(1):e01477–18.
Kargarpour Kamakoli M, Hadifar S, Khanipour S, Farmanfarmaei G, Fateh A, Siadat SD, et al. Comparison of MIRU-VNTR genoty** between old and fresh clinical samples in tuberculosis. Infect Dis. 2019;51(9):659–67.
Kamakoli MK, Farmanfarmaei G, Masoumi M, Khanipour S, Gharibzadeh S, Sola C, et al. Prediction of the hidden genotype of mixed infection strains in Iranian tuberculosis patients. Int J Infect Dis. 2020;95:22–7.
Von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med. 2007;147(8):573–7.
Stijnen T, Hamza TH, Özdemir P. Random effects meta-analysis of event outcome in the framework of the generalized linear mixed model with applications in sparse data. Stat Med. 2010;29(29):3046–67.
Huedo-Medina TB, Sánchez-Meca J, Marín-Martínez F, Botella J. Assessing heterogeneity in meta-analysis: Q statistic or I2 index? Psychol Methods. 2006;11(2):193.
Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36(3):1–48.
Egger M, Smith GD, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. Bmj. 1997;315(7109):629–34.
Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, et al. High functional diversity in mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008;6(12):e311.
Couvin D, Reynaud Y, Rastogi N. Two tales: worldwide distribution of central Asian (CAS) versus ancestral east-African Indian (EAI) lineages of mycobacterium tuberculosis underlines a remarkable cleavage for phylogeographical, epidemiological and demographical characteristics. PLoS One. 2019;14(7):e0219706.
Tarashi S, Fateh A, Jamnani FR, Siadat SD, Vaziri F. Prevalence of Bei**g and Haarlem genotypes among multidrug-resistant mycobacterium tuberculosis in Iran: systematic review and meta-analysis. Tuberculosis. 2017;107:31–7.
Mokrousov I, Shitikov E, Skiba Y, Kolchenko S, Chernyaeva E, Vyazovaya A. Emerging peak on the phylogeographic landscape of mycobacterium tuberculosis in West Asia: definitely smoke, likely fire. Mol Phylogenet Evol. 2017;116:202–12.
Acknowledgments
None to disclose.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conception and design: SH, SM and FV. Search strategy: SH and SM. Study selection: SH and AF. Data extraction: SH and AF. Data synthesis and analysis: VP and SM. Data interpretation: SH, SM and FV. Manuscript drafting: SH, SM, SDS and FV. Manuscript revision: SDS, SM and FV. Approved the final version of the manuscript: All authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicting interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hadifar, S., Fateh, A., Pourbarkhordar, V. et al. Variation in Mycobacterium tuberculosis population structure in Iran: a systemic review and meta-analysis. BMC Infect Dis 21, 2 (2021). https://doi.org/10.1186/s12879-020-05639-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-020-05639-7