
Abstract
Background
Heterosis breeding is one of the most important breeding methods for chrysanthemum. To date, the genetic mechanisms of heterosis for waterlogging tolerance in chrysanthemum are still unclear. This study aims to analyze the expression profiles and potential heterosis-related genes of two hybrid lines and their parents with extreme differences in waterlogging tolerance under control and waterlogging stress conditions by RNA-seq.
Results
A population of 140 F1 progeny derived from Chrysanthemum indicum (Nanchang) (waterlogging-tolerant) and Chrysanthemum indicum (Nan**g) (waterlogging-sensitive) was used to characterize the extent of genetic variation in terms of seven waterlogging tolerance-related traits across two years. Lines 98 and 95, respectively displaying positive and negative overdominance heterosis for the waterlogging tolerance traits together with their parents under control and waterlogging stress conditions, were used for RNA-seq. In consequence, the maximal number of differentially expressed genes (DEGs) occurred in line 98. Gene ontology (GO) enrichment analysis revealed multiple stress-related biological processes for the common up-regulated genes. Line 98 had a significant increase in non-additive genes under waterlogging stress, with transgressive up-regulation and paternal-expression dominant patterns being the major gene expression profiles. Further, GO analysis identified 55 and 95 transgressive up-regulation genes that overlapped with the up-regulated genes shared by two parents in terms of responses to stress and stimulus, respectively. 6,640 genes in total displaying maternal-expression dominance patterns were observed in line 95. In addition, 16 key candidate genes, including SAP12, DOX1, and ERF017 which might be of significant importance for the formation of waterlogging tolerance heterosis in line 98, were highlighted.
Conclusion
The current study provides a comprehensive overview of the root transcriptomes among F1 hybrids and their parents under waterlogging stress. These findings lay the foundation for further studies on molecular mechanisms underlying chrysanthemum heterosis on waterlogging tolerance.
Similar content being viewed by others


Heterosis refers to the phenomenon where hybrids exhibit superior traits compared to their parents in agronomic characteristics [1]. Heterosis breeding has been proposed and applied for over a century, but there is still no complete and clear explanation for the formation mechanism of heterosis [2]. The overdominance theory proposes that heterosis arises from favorable interactions between alleles in heterozygotes, leading to heterozygotes performing better than homozygotes [3, 4]. Overdominant effects have been observed in various heterosis traits in different plants, such as rice [5], maize [6], and tomato [7]. The dominance theory suggests that heterosis comes from the mask of unfavorable recessive genes by favorable dominant genes at heterozygous loci [8, 9]. This theory has found evidence in many hybrid species of diploid plants [10, 11]. The epistasis theory suggests that the interaction between multiple alleles controlling major traits gives rise to multiplicative effects, and these genes controlling sub-traits collectively contribute to heterosis [12]. Evidence of epistasis involvement in heterosis has been found in rice [13, 14] and maize [15, 16]. The dosage balance in gene regulation hypothesis proposes that the dosage effects of non-additive genes modulate heterosis by affecting dosage-dependent regulatory systems, with transcription factors, chromatin proteins, and signaling transduction cascade members being the major factors of dosage-dependent regulatory systems [17,18,19]. Recent studies suggest that it is challenging to identify common genomic regions that explain heterosis in different hybrid combinations. Therefore, focusing on the analysis of gene expression patterns may provide a better understanding of heterosis[20]. These mechanisms described by different theories can not be fully applied to every heterosis trait. The formation mechanisms of heterosis for some traits may also involve multiple effects [2, 21], as the phenomenon of heterosis is based on the non-linear effects of multiple heterozygous gene combinations of parental genetic differences [12, 22], which also restrict individual exploration of heterosis from a genetic perspective.
Due to potential interactions within the parental genomes, the gene expression patterns and levels of hybrids are often taken as a focus in dissecting heterosis. For example, it was found that the yield heterosis might be associated with genes related to energy metabolism and sugar metabolism pathways among differentially expressed genes (DEGs), according to RNA-seq performed on rice hybrids at the panicle primordium differentiation stage and grain filling stage [23]. In another transcriptome analysis of the hybrid rice ** of waterlogging tolerance in chrysanthemum. Planta. 2016;244(6):1241–52." href="/article/10.1186/s12870-024-04954-4#ref-CR36" id="ref-link-section-d254893368e686">36]. In analyzing the interrelationships among combining ability, genetic distance, and heterosis using a 4 × 3 incomplete diallel cross, it was found that six growth and biomass-related traits were mainly controlled by non-additive gene effects [37]. By constructing a genetic linkage map using 162 F1 progeny, 37 unconditional QTLs, 51 conditional QTLs, and several epistasis QTLs associated with waterlogging tolerance were successfully identified [38]. Several wild Chrysanthemum species give access to resistance breeding by providing pivotal candidate resistance genes that are not usually absent in cultivated chrysanthemum [34, 39]. Wang et al. (2013) obtained hybrid offspring with stronger waterlogging tolerance through interspecific hybridization between C. morifolium cv. ‘Nannong Yinshan’ and C. zawadskii, demonstrating that interspecific hybridization is an effective way to improve the waterlogging tolerance of cultivated chrysanthemums [40]. To date, little is known about the mechanisms of heterosis on waterlogging tolerance, and the causal genes responsible for waterlogging tolerance urgently need to be dissected in chrysanthemums. Chrysanthemum indicum has been regarded as making significant contributions to the origin of cultivated chrysanthemum, which has wide geographical distribution, multiple ecotypes, and high genetic diversity, severing a model species for hexaploidy chrysanthemum [41].
In this study, we assessed the genetic variation and heterosis of waterlogging tolerance in an F1 population derived from waterlogging-tolerant C. indicum (Nanchang) and waterlogging-sensitive C. indicum (Nan**g) [42]. To further explore the formation mechanism of waterlogging tolerance heterosis in chrysanthemums, two hybrid offspring, lines 98 and 95, exhibiting extreme differences in waterlogging tolerance, as well as two parents, were selected to investigate the global transcriptomes before and after waterlogging stress using RNA-seq. Differentially expressed transcripts and their expression patterns were analyzed, and 16 key candidate genes assigned to GO terms for response to stress and hormone response were identified. Our findings provide new insights into the mechanisms underlying waterlogging tolerance heterosis in chrysanthemums and contribute to the utilization of heterosis in future breeding programs.
Materials and methods
Materials
The experimental materials consisted of the highly waterlogging-tolerant maternal parent, C. indicum (Nanchang) (termed NC), and the waterlogging-sensitive paternal parent, C. indicum (Nan**g) (termed NJ), along with 140 F1 lines. Hybridization was conducted from October to November 2018, with the maternal plants bagged immediately after emasculation and artificial pollination. Mature and fully developed seeds were harvested two months after pollination and stored at room temperature. In March 2019, the seeds were sown in polystyrene foam trays containing a substrate mixture of vermiculite and perlite (3:1 ratio). After germination, which took approximately 60 days, the seedlings were transplanted into the field and sequentially numbered using Arabic numerals. Two rounds of top pruning were performed on the seedlings to generate sufficient cuttings for waterlogging tolerance identification. All the materials were preserved at the Chrysanthemum Germplasm Resource Preservation Center of Nan**g Agricultural University (118.985°N, 32.068°E).
Waterlogging treatment and phenotypic evaluation
The potted waterlogging experiments were conducted in a greenhouse from August to October 2019 and June to August 2020. Healthy, uniform plants of two parents and 140 F1 progeny were selected for the experiments. The 6 cm cuttings were planted in 32-hole high-footed plug trays filled with a sterile substrate mixture of perlite and vermiculite in a ratio of 1:3. Once the cuttings developed roots and reached approximately ten leaves, they were transferred to turnover boxes (65 cm × 43 cm × 16 cm) for further treatment. The experiments consisted of control and treatment groups. For both the parents and their offspring, the treatment groups were replicated 10 plants per individual, while the control groups were replicated 6 plants per individual. The waterlogging stress was imposed by maintaining 3 cm above the soil surface, and water was supplied regularly to maintain the stress level [36, 43]. The control plants were subjected to normal watering management. The temperature during the two experimental periods was controlled at 20–25°C, with a relative humidity of 80% and a photoperiod of 16 h.
After 9 days of waterlogging treatment, scores were recorded based on four morphological indicators: leaf color (1–7 points), leaf shape (1–5 points), stem color (1–4 points), and stem shape (1–4 points) to assess the appearance of the plants following waterlogging. Since the control group exhibited good growth, the Score was only performed on the treatment group. The score criteria were chosen according to the method described in a previous study [36], where the score values were derived from a combination of four morphological indicators. Six growth parameters, i.e., shoot height (SH), root length (RL), shoot fresh weight (SFW), shoot dry weight (SDW), root fresh weight (RFW), and root dry weight (RDW), were measured for both the control and treatment groups. The measurements of these growth parameters were taken immediately after the ending of stress treatment. SH and RL were measured using a ruler, while SFW and RFW were measured using an electronic balance. Then, the samples were placed in envelopes and dried at 105°C until a constant weight was reached. SDW and RDW were measured using an electronic balance. The average of ten replicates were used as the phenotypic values of each individual for the Score and 6 biomass traits.
To describe the differences in waterlogging tolerance among the parents and 140 F1 lines, the waterlogging tolerance index (WI = treatment group value/control group value) was calculated for each measured trait. Furthermore, the waterlogging tolerance of each entry in C. indicum F1 population was evaluated using the membership function method via the following formula: Xi = (X − Xmin) / (Xmax − Xmin) × 100%, where Xi represents the membership function value of the i-th traits, X represents the waterlogging tolerance index value of the i-th traits, and Xmax and Xmin represent the maximum and minimum values of the waterlogging tolerance traits for the i-th traits, respectively. The average membership function values of waterlogging tolerance traits (MFVW) were used to represent the waterlogging tolerance of each entry, where a higher MFVW indicates more tolerance. Following the approach by Yan [44], the waterlogging tolerance grade of the F1 population was classified into five categories based on the MFVW and standard deviation (SD): highly waterlogging-tolerant (HWT, Xi ≥ MFVW + 1.64SD), waterlogging-tolerant (WT, MFVW + 1SD ≤ Xi ˂ MFVW + 1.64SD), moderately waterlogging-tolerant (MWT, MFVW − 1SD ≤ Xi ˂ MFVW + 1SD), waterlogging-sensitive (WS, MFVW − 1.64SD ≤ Xi ˂ MFVW − 1SD), and highly waterlogging-sensitive (HWS, Xi ˂ MFVW − 1.64SD), where Xi represents the membership function value of the i-th hybrid line.
Phenotypic data processing and analysis
The calculation of heterosis was based on the method described by Zhang et al. [45], using two parameters: mid-parent heterosis (MPH) and high-parent heterosis (HPH). Data input and organization were performed using Microsoft Excel 2019 software. Descriptive statistical analysis, correlation analysis, and significance analysis of heterosis were conducted using SPSS 24 software.
Sample preparation and RNA sequencing
Root samples were collected from two parental plants, C. indicum (Nan**g) and C. indicum (Nanchang), as well as two F1 lines with extreme differences in waterlogging tolerance, lines 98 (HWT) and 95 (HWS), after 0 h and 12 h waterlogging treatment. Three biological replicates were prepared, with each replicate consisting of three consistent plants. Approximately 0.5 g of each sample was collected after mixed-grinding, rapidly frozen in liquid nitrogen, and then stored at -80°C. Total RNA was extracted using the Trizol reagent kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The RNA quality was evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and RNase-free agarose gel electrophoresis was performed to confirm the integrity of the RNA. After total RNA extraction, the rRNA was removed using the Ribo ZeroTM Magnetic Kit (Epicentre, Madison, WI, USA) to enrich for mRNA, which was then reverse-transcribed into cDNA using random primers. The cDNA fragments were purified using the QiaQuick PCR Purification Kit (Qiagen, Venlo, the Netherlands), followed by end repair, the addition of A bases, and ligation to Illumina sequencing adapters. Gel electrophoresis was performed to select the appropriate size range of the ligated products, followed by PCR amplification. Sequencing was conducted using the Illumina NovaSeq 6000 platform (Gene Denovo Biotechnology Co., Guangzhou, China).
Sequence assembly and data analysis
The raw reads obtained from sequencing were subjected to quality control using the fastp software [46]. This process involved the removal of reads containing adapters and reads consisting entirely of A bases. Additionally, low-quality data (more than 50% base quality value Q ≤ 20) was filtered. This resulted in the generation of clean reads. The Trinity software was used for read assembly [47]. Trinity first merged reads with a certain length overlap to generate longer fragments. These assembled fragments, obtained through read overlap relationships and devoid of N bases, were considered assembled unigenes. The quality of the assembly results was evaluated using metrics such as the N50 value, sequence length, and Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis (http://busco.ezlab.org/). To annotate the unigene sequences, a blastx search was performed against protein databases including NR, SwissProt, KEGG, and COG/KOG, with an E-value cutoff of < 1E-5. This allowed the identification of the protein with the highest sequence similarity to the given unigene, providing protein functional annotation information for the unigene. For gene differential expression analysis, the input data consisted of read counts obtained from gene expression level analysis. The edgeR software was utilized for differential analysis between samples [48], while the DESeq2 software was employed for differential analysis between groups [49]. Genes with a false discovery rate (FDR) ≤ 0.05 and a fold change ≥ 2 were selected as significant DEGs. The assembled unigenes were quantified using the RSEM software [50], and gene expression levels were calculated using the reads per kilobase of transcript per million mapped reads (RPKM) method. Transcriptome sequencing and analysis were conducted by Guangzhou Jidio Biotechnology Co., Ltd. Venn diagrams and heatmaps of gene expression levels were generated using the omicsmart platform tools.
Quantitative real-time PCR validation of RNA sequencing
Six DEGs were randomly selected to validate the reliability of RNA sequencing data. Primers for gene validation were designed using Primer 5.0 software. The reference gene, the EF1α gene of C. indicum, was selected for normalization. The qRT-PCR procedure followed the methods described by Ren et al. [51]. Each sample was subjected to three technical replicates. The primer sequences used are listed in Table S1.
GO and KEGG pathway enrichment analysis
The DEGs were mapped to various terms in the Gene Ontology (GO) database (http://www.geneontology.org/), and the number of genes associated with each term was calculated. This allowed for the identification of gene lists and the statistical determination of the number of genes associated with specific GO functions. GO terms that met the threshold of Q value ≤ 0.05 were defined as significantly enriched terms. Pathway analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) web server (http://www.kegg.jp/) [52, 53]. After multiple testing corrections, pathways with a Q value ≤ 0.05 were defined as significantly enriched pathways.
Classification and identification of gene expression patterns
The average expression values of three biological replicates of two hybrids were compared with the mid-parent values after 0 h and 12 h of waterlogging stress, respectively. Short Time-series Expression Miner software was used to analyze the gene expression patterns [54]. The classification of gene expression patterns in hybrids was based on the study by Swanson-Wagner et al. [55]. Genes were categorized into eight significant types (Q-value ≤ 0.05) based on the difference in expression levels between hybrids and parents, i.e., additive (hybrid gene expression equal to the average of parents), two types of overdominance (hybrid gene expression significantly higher or lower than parents), two types of high-parent dominance, and two types of low-parent dominance (hybrid gene expression equal to one parent and significantly higher or lower than the other parent).
Results
Waterlogging tolerance identification of C. indicum F1 progeny
Statistical analysis of the F1 population after 9 days of waterlogging stress revealed a wide range of phenotypic variations among the seven investigated traits (Table S2, Fig. 1a-g). The coefficients of variation ranged from 14.47 to 59.38%, with SFW and SDW showing higher variability than other traits, indicating a more pronounced response of the root system to waterlogging stress. Based on their MFVWs, 68% of the F1 progeny were classified as moderately waterlogging-tolerant, while 7 and 6 individuals belonged to the HWT and HWS categories, accounting for 5.63% and 4.93%, respectively (Table S3, Fig. 1h). The phenotypes of five representative F1 lines after 9 days of treatment are shown in Fig. S1. Heterosis analysis revealed significant positive mid-parent heterosis in the F1 population for score, RL, SFW, SDW, and RDW, ranging from 10.09 to 34.41%. MFVW has the weakest positive mid-parent heterosis (2.93%) and is insignificant. The high-parent heterosis for all waterlogging tolerance traits in this F1 population was negative (Table 1). However, several individuals with positive and negative transgressive segregation were found for these traits (Fig. 1), indicating a significant transgressive segregation phenomenon in the F1 generation.

Frequency distribution of seven waterlogging tolerance traits and box plot of MFVW in the F1 population derived from C. indicum (Nanchang) and C. indicum (Nan**g). a Shoot height. b Root length. c Shoot fresh weight. d Root fresh weight. e Shoot dry weight. f Root dry weight. g Score. The green and orange arrowheads in panel a-g indicate the two parents: NC, C. indicum (Nanchang); NJ, C. indicum (Nan**g). h Box plot showing the variation of MFVWs among the five waterlogging tolerance levels in F1 lines
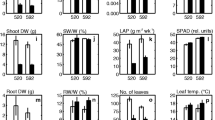
Notably, line 95 had obvious yellowing, wilting, and even rotting of the lower leaves, and its root mass was significantly reduced compared to the control group. By contrast, line 98 showed no significant morphological changes (Fig. 2a). Heterosis analysis revealed that line 98 exhibited positive mid-parent heterosis for all waterlogging tolerance traits (Table S4, Fig. 2b-g), with Score showing the maximal value (58.73%), followed by RDW (32.56%). Additionally, line 98 displayed positive high-parent heterosis for all traits except RFW and RL, and the maximal high-parent heterosis rate was observed in Score (5.26%), indicating that line 98 behaved stronger waterlogging tolerance compared to the waterlogging-tolerant parent C. indicum (Nanchang). In contrast, line 95 had the lowest MFVW in this F1 population and exhibited negative mid-parent heterosis and high-parent heterosis for all traits (Table S4, Fig. 2b-g), indicating its weaker waterlogging tolerance than the waterlogging-sensitive parent C. indicum (Nan**g). Therefore, lines 98 and 95 were selected to study waterlogging tolerance heterosis further.

Phenotypic characteristics of F1 hybrids 95 and 98 and their parents. a plant phenotype of hybrids and parents after 9 days of waterlogging stress. Scale bar = 2 cm. b-g Statistical analysis of shoot height (SH), root length (RL), shoot fresh weight (SFW), root fresh weight (RFW), shoot dry weight (SDW), and root dry weight (RDW) of F1 hybrid 95 and 98 and their parents. Different letters indicate significant difference at P < 0.05 between individuals. Data represent mean values ± SE. The control group is prefixed with C; The 9-day waterlogging stress treatment group is prefixed with W; NC indicates female parent C. indicum (Nanchang), and NJ indicates male parent C. indicum (Nan**g)
RNA sequencing and quantitative real-time PCR validation
To study the differences in the response mechanisms to waterlogging stress between hybrid lines and parents, RNA of root tissues was extracted to construct 24 libraries. As a result, 47.94–54.93 million raw reads were yielded for each sample. After filtering the raw read data, we obtained 46.7–54.0 million clean reads, corresponding to 6.97–8.06 Gb of clean bases. The Q30 levels ranged from 91.87 to 93.05%, and the GC content ranged from 41.69 to 44.63%. A total of 119,965 unigenes were assembled, with lengths ranging from 201 to 1,446 bp and an average length of 808 bp. The N50 value was greater than 1,264 bp. Based on the annotation results from the Nr, KEGG, KOG, and SwissProt databases, a total of 61,549 unigenes were annotated in at least one database. The average Pearson’s correlation coefficient among the three samples was 0.9675 (Fig. S2), indicating that the transcript abundances of the biologically replicated samples were highly correlated. These results suggest the high quality of RNA-seq. Detailed information about the RNA-Seq data was provided in Table S5.
The reliability of the RNA-seq data was further validated using the qRT-PCR method. Six genes were randomly selected for validation, including AP2-ERF transcription factor (Unigene21365), late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein family (Unigene101624), DCD (Development and Cell Death) domain protein (Unigene43272), lipoxygenase 1 (Unigene3146), peroxidase superfamily protein (Unigene22039) and abscisic acid receptor PYL4-like (Unigene77240). As a result, all of the six genes examined showed similar expression profiles to the RNA-seq data (Fig. S3), confirming the reliability of the RNA-seq data.
Identification of DEGs under waterlogging stress
Sixteen pairwise comparisons (C-98 vs. W-98, C-95 vs. W-95, C-NC vs. W-NC, C-NJ vs. W-NJ/C-NC vs. C-98, C-NJ vs. C-98, C-NJ vs. C-NC/W-NC vs. W-98, W-NJ vs. W-98, W-NJ vs. W-NC/C-NC vs. C-95, C-NJ vs. C-95, C-NJ vs. C-NC/W-NC vs. W-95, W-NJ vs. W-95, W-NJ vs. W-NC) were made to examine the DEGs (fold change ≥ 2 and FDR ≤ 0.05). In the comparisons between the control and waterlogging stress groups, a total of 15,572 (7,546 up-regulated and 8,026 down-regulated), 10,763 (5,233 up-regulated and 5,530 down-regulated), 10,931 (5,541 up-regulated and 5,390 down-regulated), and 14,810 (7,395 up-regulated and 7,415 down-regulated) DEGs were identified for line 98, line 95, NC, and NJ, respectively. Compared to the parental and highly waterlogging-sensitive line 95, the highly waterlogging-tolerant hybrid line 98 exhibited the maximal number of DEGs, indicating its strong response to waterlogging stress (Fig. 3a, b).In the pairwise comparisons of C-NC vs. C-98, C-NJ vs. C-98, and C-NJ vs. C-NC, 15,245 (12,348 up-regulated and 2,897 down-regulated), 13,965 (8,180 up-regulated and 5,785 down-regulated), and 23,132 (7,478 up-regulated and 15,654 down-regulated) DEGs were identified, respectively (Fig. 3c-e). In the pairwise comparisons of W-NC vs. W-98, W-NJ vs. W-98, and W-NJ vs. W-NC, 17,050 (13,373 up-regulated and 3,677 down-regulated), 21,582 (13,203 up-regulated and 8,379 down-regulated), and 24,844 DEGs (8,605 up-regulated and 16,239 down-regulated) were identified, respectively (Fig. 3f-h). In addition, more parental DEGs were discovered in line 98 under waterlogging stress, indicating line 98 might have different response mechanisms to waterlogging stress from those of its parents, and the related DEGs might accordingly explain the waterlogging tolerance heterosis.

Basic analysis of differentially expressed genes. a, b Histogram and venn diagrams showing the number of up-regulated and down-regulated differentially expressed genes among hybrids 95 and 98 and their parents under waterlogging stress and control conditions. c-e Histogram and venn diagrams of up-regulated and down-regulated differentially expressed genes in paired comparison between hybrids and parents under control condition. f-h Histogram and venn diagrams of up-regulated and down-regulated differentially expressed genes in paired comparison between hybrids and parents under waterlogging stress condition. The control group is prefixed with a capital letter C; The 9-day waterlogging stress treatment group is prefixed with a capital letter W; NC, C. indicum (Nanchang); NJ, C. indicum (Nan**g)
Correlations between the hybrid and its parents in gene expression were investigated with cluster analysis using Cluster 3.0 software. The transcriptome profiles of line 95 were similar to C. indicum (Nan**g) under waterlogging stress and control conditions. The transcriptome profiles of line 98 were identical to C. indicum (Nan**g) (male parent) under control conditions, but it was more similar to C. indicum (Nanchang) (female parent) under waterlogging stress conditions (Fig. S4).
Functional enrichment analysis of the common DEGs under waterlogging stress
In the pairwise comparisons between the control group and the waterlogging stress group, 3,931 genes (1,720 up-regulated, 2,132 down-regulated) out of the total 15,572 DEGs were found to be in common among line 98 and the parents (Fig. 4a, b). The functional enrichment analysis uncovered that these common genes were involved in 55 biological processes (FDR ≤ 0.05), including several terms related to response to stimulus (GO:0050896), hormone (GO:0009725), stress (GO:0006950), etc. (Table S6). Further analysis of the common up-regulated genes showed that 63 GO biological processes were significantly enriched (Table S7), including various processes involved in plant responses to abiotic stress, particularly responses to stimulus (GO:0050896) and stress (GO:0006950), with 451 and 295 genes significantly enriched, respectively. Additionally, terms related to ethylene metabolism and hormone response, such as ethylene metabolic process (GO:0009692), cellular alkene metabolic process (GO:0043449), and response to hormone (GO:0009725), were also enriched. As for the common down-regulated genes, 45 GO biological process terms were significantly enriched, including processes related to RNA modification (GO:0009451), ribonucleoprotein complex biogenesis (GO:0022613), and organic substance transport (GO:0071702) (Table S8). The functional enrichment analysis of the common DEGs between line 98 and the waterlogging-tolerant parent C. indicum (Nanchang) revealed 14 significant biological process terms, such as phytoalexin biosynthetic process (GO:0052315), indole phytoalexin metabolic process (GO:0046217), and phytoalexin metabolic process (GO:0052314). However, no significant biological process terms were enriched in the specific DEGs of line 98 or its common DEGs with the waterlogging-sensitive parent C. indicum (Nan**g).

Basic analysis of differentially expressed genes and KEGG enrichment analysis of line 98 and its parents under waterlogging stress and control conditions. a, b Venn diagram of up-regulated and down-regulated differentially expressed genes. c KEGG enrichment map of 3,931 common DEGs of line 98. The numbers on the right side of the bar graph represent the number of genes contained in the corresponding KEGG pathway; The numbers in parentheses represent the significance level of the corresponding KEGG pathway (-Log10(Q value)). The KEGG enrichment map mentioned in the figure has been granted copyright permission by Kanehisa Laboratories
In the meantime, KEGG pathway enrichment analysis was conducted against the 3,931 common DEGs. The results revealed that 12 KEGG pathways involving a total of 827 DEGs, including phenylpropanoid biosynthesis, ribosome, biosynthesis of unsaturated fatty acids, metabolic pathways, stilbenoid, diarylheptanoid, and gingerol biosynthesis, α-linolenic acid metabolism, ribosome biogenesis in eukaryotes, and biosynthesis of secondary metabolites, etc., were significant enriched (Fig. 4c).
Expression pattern analysis of DEGs between hybrids and parents
Based on the expression profiles of parents and hybrids, gene expression levels were categorized into eight groups, i.e., Profile 0 - Profile 7 (Fig. 5a-d). Profile 0 and Profile 7 represented additive expression, Profile 1, Profile 3, Profile 4, and Profile 6 represented dominant expression, and Profile 2 and Profile 5 represented overdominant expression, respectively. Trend analysis was performed using the DEGs between hybrids and parents under control and waterlogging stress conditions. Intriguingly, four expression patterns, namely, additivity (Profile 0), transgressive up-regulation (Profile 5), paternal-expression dominance (Profile 3), and maternal-expression dominance (Profile 1), were significantly enriched for line 98 (Q value ≤ 0.05) under control and waterlogging stress conditions. Additionally, line 98 displayed an enrichment of maternal-expression dominance pattern (Profile 6) under waterlogging stress. These non-additive expression patterns contained a total of 25,829 genes in line 98 under waterlogging stress, accounting for 39.41% of the total DEGs (65,547), which was much higher than that in line 95 (14,813 DEGs; 21.63%). Under control condition, line 98 showed the maximal number of DEGs in Profile 3 (9,918), followed by Profile 5 (4,680) (Fig. 5e). However, under waterlogging stress, the two maternal-expression dominance patterns (5,204 for Profile 6 and 4,417 for Profile 1) and overdominant expression pattern (7,148 for Profile 5) in line 98 possessed higher number of DEGs (Fig. 5b, e). These results suggested that the three non-additive expression patterns may play an important role in waterlogging tolerance heterosis in chrysanthemums, especially the overdominant expression pattern with the highest increase in genes after experiencing waterlogging stress. Under both control and waterlogging stress conditions, the highest number of DEGs in maternal expression dominance patterns was observed in line 95 (Fig. 5c, e). However, after being subjected to waterlogging stress, the number of genes was decreased in line 95 for all expression patterns except profile 7 (Fig. 5d, e).

Analysis of gene expression trends in hybrids compared to parents under control and waterlogging stress conditions. a, b The gene expression genetic patterns of line 98. c, d The gene expression genetic patterns of line 95. The corresponding P-value is marked in the expression pattern diagram, and the significant expression patterns (P < 0.05) are marked in gray. e Statistical analysis of the number of genes contained in each expression pattern. The control group is prefixed with a capital letter C; The 9days waterlogging stress treatment group is prefixed with a capital letter W; the numbers after C and W represent biological repeated numbers; NC, C. indicum (Nanchang); NJ, C. indicum (Nan**g)
Comparative and functional enrichment analyses of non-additive genes in hybrids
To further investigate the waterlogging tolerance heterosis in line 98, enrichment analyses of the DEGs showing transgressive upregulation (profile5) and the maternal expression dominance (profile6) under waterlogging stress were conducted. The results revealed that the DEGs in profile 5 were enriched in 189 GO biological process terms, of which some were related to waterlogging tolerance, including responses to osmotic stress (GO:0006970), response to stress (GO:0006950), stimulus (GO:0050896), and hormone (GO:0009725) (Table S9). However, the DEGs in Profiles 1 and 6 did not show significant enrichment in biological process terms. Under waterlogging stress condition, 5,069 out of the 7,148 transgressive up-regulation genes in line 98 were specific to waterlogging stress, which was assigned to 148 GO biological process terms with significant enrichment in waterlogging tolerance-related GO terms, including responses to stress (GO:0006950), stimulus (GO:0050896), osmotic stress (GO:0006970), hormone (GO:0009725), and external stimuli (GO:0009605). The top 20 significantly enriched GO terms and KEGG pathways are shown in Fig. 6a and b. The response to stress term contained 747 genes with an overlap of 55 genes within the 1,720 common up-regulated DEGs (Table S10). The response to the stimulus term contained 1,179 genes, with an overlap of line 95 genes within the common up-regulated DEGs. Furthermore, many stress-related genes were found among these overlapped genes, such as PUB23-like (Unigene0065212), WRKY17 (Unigene0076621), RHA2A (Unigene0020876), and SAP12 (Unigene0072913), as well as three genes involved in reactive oxygen species defense: DOX1 (Unigene0031048), PER64 (Unigene0124910), and PER47 (Unigene0022039). The heatmap of expression levels demonstrated that the above genes exhibited higher expression levels in line 98 compared to that in line 95 and two parents, indicating their potential roles in the waterlogging tolerance heterosis (Fig. 6c). Notably, SAP12 and DOX1 showed a 12-fold and 10-fold up-regulation under waterlogging treatment, respectively. Among the 120 genes covered by the response to hormone (GO:0009725) term, five transcription factors related to ethylene signaling were identified, including ERF008 (Unigene0040439), ERF1A (Unigene0126404), ERF1B (Unigene0021365), ERF9 (Unigene0061735), and ERF017 (Unigene0095957). Specifically, ERF017 and ERF1B showed 38-fold and 7-fold up-regulation under waterlogging stress, respectively. In addition to ethylene, several enriched DEGs related to other hormones were also discovered, such as auxin-responsive factor ARF19 (Unigene0001119), jasmonic acid signaling-related transcription factor MYC2 (Unigene0067836), abscisic acid receptor PYL4-like (Unigene0077240), and brassinosteroid-associated gene BAK1 (Unigene0000389). These hormone-responsive genes also exhibited significantly higher expression levels in line 98 (Fig. 6d), indicating their potential contribution to waterlogging tolerance. Overall, through RNA-seq, we provided functional links of heterosis to waterlogging tolerance in C. indicum. Nevertheless, these potential genes still need more evidence to support them in further detail.

Enrichment analysis of 5,069 unique transgressive up–regulation genes and expression heat map of 16 candidate genes. a Enrichment of GO biological process terms for 5,069 unique transgressive up–regulation genes of line 98. Only the top 20 GO terms with the lowest Q value are shown. b KEGG enrichment of 5,069 unique transgressive up–regulation expression genes in line 98. Only the top 20 pathways with the lowest Q value are shown. The numbers on the right side of the bar graph represent the number of genes contained in the corresponding GO terms. The numbers in parentheses represent the significance level of the corresponding GO terms (-Log10(Q value)). The KEGG enrichment map mentioned in the figure has been granted copyright permission by Kanehisa Laboratories. c Expression heatmaps of 9 hormone response-related genes enriched in GO terms in response to the hormone. d Expression heatmaps of 7 plant stress resistance-related genes enriched in GO terms in response to stress. The legend at the right of the figure indicates the gene expression levels transformed by log10 (FPKM)
Discussion
The utilization of heterosis has long been one of the main objectives of chrysanthemum breeders. Although heterosis has been widely exploited in improving the tolerance to biotic and abiotic stresses of cultivated chrysanthemum via intraspecific and interspecific crosses [40, 56], the molecular and genetic mechanisms underlying the phenomenon remain largely unknown, which might be attributed to the complicated genetic background and high heterozygosity of cultivated chrysanthemum. C. indicum is an ideal model species of hexaploidy chrysanthemum owing to its high phenotypic diversity and close relationship with chrysanthemum [35]. Liu et al. (2012) proposed that C. indicum (Nan**g) played an important role in the origin of cultivated chrysanthemum according to the phylogenetic analysis of single-copy nuclear gene and chloroplast DNA sequences [57]. A recent study also revealed that C. indicum (Nan**g) is more closely related to cultivated chrysanthemum based on whole genome resequencing [36]. In addition, heterosis is strongly correlated with the genetic distance between parents in studies on rice [38].
Using transcriptome sequencing to analyze DEGs between hybrids and parents is an effective way to explore possible molecular mechanisms for heterosis. In the pairwise comparisons between C. indicum (Nan**g), C. indicum (Nanchang), and line 98, it was found that the maximum number of DEGs was detected between the two parents under both control and waterlogging stress (23,132 and 24,844). There were fewer DEGs between line 98 and C. indicum (Nan**g) (13,965) under control conditions, while fewer DEGs between line 98 and C. indicum (Nanchang) (17,050) (Fig. 3c, f) were observed under waterlogging stress. This result was consistent with the clustering analysis of all transcriptions, in which line 98 was closely clustered with C. indicum (Nan**g) under control conditions, whereas it was separated under stress (Fig. S4). This phenomenon of genes in hybrids tending to be biased towards specific parents in their expression has also been reported in maize, Arabidopsis, and rice [61,62,−63]. The interaction between waterlogging-responsive genes inherited from different parents in line 98 might activate or suppress more responsible genes. Additionally, stress-tolerance genes inherited from C. indicum (Nanchang) may exhibit distinct expression patterns in hybrids. These issues might contribute to the waterlogging tolerance heterosis observed in line 98.
Reasonable gene selection and discovery are critical aspects of transcriptome analysis. To identify waterlogging-tolerant-related DEGs, three pairwise comparisons (C-98 vs. W-98, C-NC vs. W-NC, C-NJ vs. W-NJ) were performed. A total of 3,931 common DEGs and line 98 specific genes were identified in this study (Fig. 3a, b). GO enrichment analysis demonstrated that these parental common genes were mainly involved in several processes directly related to plant stress responses (Table S6). However, the DEGs specific to line 98 did not enrich any biological process terms. This suggests that the common DEGs play an important role in the waterlogging tolerance heterosis of line 98, consistent with previous studies [29, 31, 32, 64] that focused on shared genes for heterosis analysis. Further enrichment analysis revealed that among the up-regulated shared genes, 295 genes were enriched in the “response to stress” (GO:0006950) term, and 451 genes were enriched in the “response to stimulus” (GO:0050896) term, which might play a crucial role in response to waterlogging stress and contribute to the waterlogging tolerance heterosis observed in line 98. This further confirms that the expression levels of common genes related to waterlogging stress response in line 98 have been changed under the combined action of the parental genomes, thereby enhancing its waterlogging tolerance.
Non-additive effects have been proven to play a vital role in the formation of plant heterosis [65]. In this study, trend analysis was conducted on the DEGs of lines 98 and 95 under control and waterlogging stress conditions. Among the five significant expression patterns identified in line 98, the number of genes assigned to the transgressive up-regulation pattern increased dramatically under waterlogging stress, followed by two maternal-expression dominant patterns (Fig. 5e). Further GO enrichment analysis revealed that the genes belonging to the transgressive up-regulation pattern were significantly enriched in multiple stress response-related biological process terms, indicating the important role of transgressive up-regulation genes in the waterlogging tolerance heterosis of chrysanthemum. Similar findings have been reported in previous studies on rice [66] and maize [67], where multiple key yield-related heterosis loci exhibited transgressive up-regulation. Studies on biomass heterosis in cotton [68] and tobacco [69] also found that the primary expression pattern of DEGs in hybrids was transgressive up-regulation. The transcriptome analysis of maize hybrid An’nong 591 also discovered that the number of overdominance genes increased more than 2.5 times under heat stress [32], which is consistent with the results of this study. GO enrichment analysis was conducted on the 5,069 waterlogging stress-specific transgressive up-regulation genes and compared them with the up-regulated parental common genes previously screened out. This further led to the identification of transgressive up-regulation parental common genes that are responsible for waterlogging stress response. The results showed that 148 GO biological process terms were significantly enriched, including responses to organic stress (GO: 0006970), endogenous stimuli (GO: 0009719), chemical (GO: 0042221), stress (GO: 0006950), and stimuli (GO: 050896). Among them, 55 genes were assigned to responses to stress terms, and 95 were assigned to responses to stimulus terms (Table S10). Furthermore, seven out of the 55 genes were selected. PUB23-like (Unigene0065212) [70] and WRKY17 (Unigene0076621) [71] have been shown to play a role in plant stress tolerance studies. RHA2A (Unigene0020876) has also been confirmed to be associated with drought and salt tolerances in previous studies on soybeans [72] and rice [73]. The expression level of the oxidative stress-related gene SAP12 (Unigene0072913) [74, 75] in super line 98 increased 12-fold after waterlogging stress treatment. DOX1 (Unigene0031048), PER64 (Unigene0124910), and PER47 (Unigene0022039) are associated with plant reactive oxygen species. The strong waterlogging tolerance of line 98 may result from the combined action of different types of stress response pathways. Ethylene plays an important role in plant response to waterlogging stress, and previous transcriptome analyses have identified ethylene-responsive genes that are important for chrysanthemum waterlogging tolerance [43]. Five transcription factors related to ethylene signaling were recognized in the waterlogging stress-specific transgressive up-regulation genes. Among them, ERF1B (Unigene0021365) and ERF017 (Unigene0095957) showed the most robust responses to waterlogging, with 7-fold and 38-fold up-regulation, respectively. These genes may also participate in the formation of waterlogging tolerance heterosis and are worth further investigating.
Conclusion
In this study, RNA-seq was used to elucidate the differences in the global gene expression profiles of C. indicum (Nan**g), C. indicum (Nanchang), as well as their positive over-dominant hybrid line 98 and negative over-dominant hybrid line 95 under control and waterlogging stress conditions. Trend analysis was also conducted to depict their gene expression patterns and explore the mechanisms underlying the waterlogging tolerance heterosis in chrysanthemums. The study ultimately highlighted the significant roles of transgressive up-regulation expressed genes and parental common up-regulated genes in the waterlogging tolerance heterosis of the hybrids. These findings provide new insights into the mechanism of heterosis and contribute to future heterosis breeding and molecular improvement of waterlogging tolerance of chrysanthemum.
Data availability
RNA-seq data of this study can be found at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/bioproject/) with the BioProject ID PRJNA1013215.
Abbreviations
- WI:
-
Waterlogging tolerance index
- MFVW:
-
Membership function value of waterlogging tolerance traits
- MPV:
-
Mid-parent value
- HPV:
-
High-parent value
- RPKM:
-
Reads per kilobase of transcript per million mapped reads
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- QTL:
-
Quantitative trait locus
- NR:
-
Non-redundant protein sequence database
- KOG:
-
Clusters of orthologous groups for eukaryotic complete genomes
- FDR:
-
False discovery rate
References
Chen Z. Genomic and epigenetic insights into the molecular bases of heterosis. Nat Rev Genet. 2013;14(7):471–82.
Swanson-Wagner R, Jia Y, DeCook R, Borsuk L, Nettleton D, Schnable P. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc Natl Acad Sci USA. 2006;103(18):6805–10.
Shull G. The composition of a field of maize. J Hered. 1908;os–4:296–301.
East E. Heterosis Genet. 1936;21:375–97.
Huang X, Yang S, Gong J, Zhao Q, Feng Q, Zhan Q, et al. Genomic architecture of heterosis for yield traits in rice. Nature. 2016;537:629–33.
Liu H, Wang Q, Chen M, Ding Y, Yang X, Liu J, et al. Genome wide identification and analysis of heterotic loci in three maize hybrids. Plant Biotechnol J. 2019;18:185–94.
Stuber C, Lincoln S, Wolff D, Helentjaris T, Lander ES. Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics. 1992;132:823–39.
Bruce A. The mendelian theory of heredity and the augmentation of vigor. Science. 1910;32(827):627–8.
James F. Alternative hypotheses of hybrid vigor. Genetics. 1948;33(5):477–87.
Li D, LU X, Zhu Y, Pan J, Zhou S, Zhang X, et al. The multi-omics basis of potato heterosis. J Integr Plant Biol. 2022;64(3):17.
Liu W, He G, Deng X. Biological pathway expression complementation contributes to biomass heterosis in Arabidopsis. Proc Natl Acad Sci USA. 2021;118(16):e2023278118.
Schnell F, Cockerham C. Multiplicative vs. arbitrary gene action in heterosis. Genetics. 1992;131:461–9.
Yu S, Li J, Xu C, Tan Y, Gao Y, Li X, et al. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA. 1997;94:9226–31.
Li H, Jiang S, Li C, Liu L, Lin Z, He H, et al. The hybrid protein interactome contributes to rice heterosis as epistatic effects. Plant J. 2020;102(1):116–28.
Tang J, Yan J, Ma X, Teng W, Wu W, Dai J, et al. Dissection of the genetic basis of heterosis in an elite maize hybrid by QTL map** in an immortalized F2 population. Theor Appl Genet. 2010;120:333–40.
Wolf D, Hallauer A. Triple testcross analysis to detect epistasis in maize. Crop Sci. 1997;37:763–70.
Liu J, Li M, Zhang Q, Wei X, Huang X. Exploring the molecular basis of heterosis for plant breeding. J Integr Plant Biol. 2019;62(3):287–98.
Birchler J, Riddle N, Auger D, Veitia R. Dosage balance in gene regulation: biological implications. Trends Genet. 2005;21(4):219–26.
Veitia B. The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol. 2010;186(1):54–62.
Birchler JA. Plant science: hybrid vigour characterized. Nature. 2016;537(7622):620–1.
Li L, Lu K, Chen Z, Mu T, Hu Z, Li X. Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics. 2008;180:1725–42.
Hochholdinger F, Hoecker N. Towards the molecular basis of heterosis. Trends Plant Sci. 2007;12(9):427–32.
Katara JL, Verma RL, Parida M, Ngangkham U, Molla KA, Barbadikar KM, et al. Differential expression of genes at panicle initiation and grain filling stages implied in heterosis of rice hybrids. Int J Mol Sci. 2020;21(1080):1–17.
Zhai R, Feng Y, Wang H, Zhan X, Shen X, Wu W, et al. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics. 2013;14(1):19.
Zhang C, Lin C, Fu F, Zhong X, Zhao L, et al. Comparative transcriptome analysis of flower heterosis in two soybean F1 hybrids by RNA-seq. PLoS ONE. 2017;12(7):e0181061.
Shahzad K, Zhang X, Guo L, Qi T, **ng C. Comparative transcriptome analysis between inbred and hybrids reveals molecular insights into yield heterosis of upland cotton. BMC Plant Biol. 2020;20:239.
Yang S, Zhang Z, Chen W, Li X, Zhou S, Liang C, et al. Integration of mRNA and miRNA profiling reveals the heterosis of three hybrid combinations of Capsicum annuum varieties. GM Crops & Food. 2021;12(1):224–41.
Xu C, Sun X, Zhang S. Mechanism on differential gene expression and heterosis formation. Hereditas. 2013;35(6):13.
Howlader J, Robin A, Natarajan S, Biswas MK, Nou IS. Transcriptome analysis by RNA–Seq reveals genes related to plant height in two sets of parent-hybrid combinations in Easter lily (Lilium longiflorum). Sci Rep. 2020;10:9082.
Li S, Jayasinghege CPA, Guo J, Zhang E, Xu Z. Comparative transcriptomic analysis of gene expression inheritance patterns associated with cabbage head heterosis. Plants. 2021;10(2):275.
Chen L, Bian J, Shi S, Yu J, He H. Genetic analysis for the grain number heterosis of a super-hybrid rice WFYT025 combination using RNA-Seq. Rice. 2018;11(1):37.
Zhao Y, Hu F, Zhang X, Wei Q, Dong J, Bo C, et al. Comparative transcriptome analysis reveals important roles of nonadditive genes in maize hybrid an’nong 591 under heat stress. BMC Plant Biol. 2019;19:273.
Bailey-Serres J, Voesenek, Laurentius AC. Flood adaptive traits and processes: an overview. New Phytol. 2015;206(1):57–73.
Yin D, Chen S, Chen F, Guan Z, Fang W. Morphological and physiological responses of two chrysanthemum cultivars differing in their tolerance to waterlogging. Environ Exp Bot. 2009;67(1):87–93.
Su J, Jiang J, Zhang F, Liu Y, Ding L, Chen S, et al. Current achievements and future prospects in the genetic breeding of chrysanthemum: a review. Hortic Res. 2019;6:109.
Su J, Zhang F, Li P, Guan Z, Fang W, Chen F. Genetic variation and association map** of waterlogging tolerance in chrysanthemum. Planta. 2016;244(6):1241–52.
Su J, Zhang F, Yang X, Feng Y, Yang X, Wu Y, et al. Combining ability, heterosis, genetic distance and their intercorrelations for waterlogging tolerance traits in chrysanthemum. Euphytica. 2017;213(2):42.
Su J, Yang X, Zhang F, Wu S, ** reveals the complex genetic architecture of waterlogging tolerance in chrysanthemum. Planta. 2018;247(4):899–924.
Song A, Su J, Wang H, Zhang Z, Zhang X, Yves V, et al. Analyses of a chromosome-scale genome assembly reveal the origin and evolution of cultivated chrysanthemum. Nat Commun. 2023;14:2021.
Wang L, Wang C, Jiang J, Chen S, Fang W, Teng N, et al. Interspecific hybridization between Chrysanthemum morifolium ‘Nannong Yinshan’ and C. Zawadskii and identification of waterlogging tolerance of their hybrid. Scientia Agricultura Sinica. 2013;46(20):4328–35.
Yang W, Glover B, Rao G, Yang J. Molecular evidence for multiple polyploidization and lineage recombination in the Chrysanthemum indicum polyploid complex (Asteraceae). New Phytol. 2006;171(4):875–86.
Su J, Yang Y, Zhang X, Li Z, Lu Z, Jia F, et al. Evaluation of wild chrysanthemums for waterlogging tolerance at the seedling stage. Euphytica. 2023;219:14.
Zhao N, Li C, Yan Y, Cao W, Song A, Wang H, et al. Comparative transcriptome analysis of waterlogging-sensitive and waterlogging-tolerant Chrysanthemum morifolium cultivars under waterlogging stress and reoxygenation conditions. Int J Mol Sci. 2018;19:1455.
Yan C, Song S, Wang W, Wang C, Sun X. Screening diverse soybean genotypes for drought tolerance by membership function value based on multiple traits and drought-tolerant coefficient of yield. BMC Plant Biol. 2020;20(1):321.
Zhang F, Chen F, Fang W, Chen S, Li F. Heterosis and mixed genetic analysis of inflorescence traits of chrysanthemum. Scientia Agricultura Sinica. 2010;43(14):2953–61.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. bioRxiv. 2018;274100.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644.
Robinson M, McCarthy D, Smyth G. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Ren L, Sun J, Chen S, Gao J, Dong B, Liu Y, et al. A transcriptomic analysis of Chrysanthemum nankingense provides insights into the basis of low temperature tolerance. BMC Genomics. 2014;15:1–20.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–51.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–92.
Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191.
Swanson-Wagner RA, Jia Y, Decook R, Borsuk LA, Nettleton D, Schnable PS. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc Natl Acad Sci USA. 2006;103(18):6805–10.
Deng Y, Chen S, Chen F, Cheng X, Zhang F. The embryo rescue derived intergeneric hybrid between chrysanthemum and Ajania przewalskii shows enhanced cold tolerance. Plant Cell Rep. 2011;(12):30.
Liu P, Wan Q, Guo Y, Yang J, Rao G. Phylogeny of the Genus Chrysanthemum L.: evidence from single-copy nuclear gene and chloroplast DNA sequences. PLoS ONE, 2012, 7.
Li D, Huang Z, Song S, **n Y, Mao D, Lv Q, et al. Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc Natl Acad Sci USA. 2016;113(41):E6026–35.
Hui L, Yuan J, Wu M, Han Z, Li L, Jiang H, et al. Transcriptome and DNA methylome reveal insights into yield heterosis in the curds of broccoli (Brassica oleracea L var. Italic). BMC Plant Biol. 2018;18:168.
Kawanabe T, Ishikura S, Miyaji N, Sasaki T, Wu L, Itabashi E, et al. Role of DNA methylation in hybrid vigor in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2016;113(43):E6704–11.
Zhang F, Ding Y, Zhang J, Tang M, Cao Y, Zhang L et al. Comparative transcriptomic reveals the molecular mechanism of maize hybrid Zhengdan538 in response to water deficit. Physiol Plant. 2022;174(6), e13818.
Dapp M, Reinders J, Bédiée A, Balsera C, Bucher E, Theiler G, et al. Heterosis and inbreeding depression of epigenetic Arabidopsis hybrids. Nat Plants. 2015;1:1–8.
Fu J, Zhang Y, Yan T, Li Y, Jiang N, Zhou Y, et al. Transcriptome profiling of two super hybrid rice provides insights into the genetic basis of heterosis. BMC Plant Biol. 2022;22:314.
Guo X, Zhang M, Zhu M, Long J, Wei Z, Li J, et al. Comparative transcriptomic analysis of the super hybrid rice Chaoyouqianhao under salt stress. BMC Plant Biol. 2022;7(1):233.
**e J, Wang W, Yang T, Zhang Q, Zhang Z, Zhu X, et al. Large-scale genomic and transcriptomic profiles of rice hybrids reveal a core mechanism underlying heterosis. Genome Biol. 2022;23:264.
Zhuang J, Fan Y, Wu J, **a Y, Zheng K. The important role of overdominant effect on rice heterosis. Sci China (Series C). 2001;31(2):106–13.
Wang B, Hou M, Shi J, Ku L, Song W, Li C, et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat Genet. 2023;55:312–23.
Li T, Wang F, Yasir M, Li K, Qin Y, Zheng J, et al. Expression patterns divergence of reciprocal F1 hybrids between Gossypium hirsutum and Gossypium barbadense reveals overdominance mediating interspecific biomass heterosis. Front Plant Sci. 2022;13:892805.
Mo Z, Luo W, Pi K, Duan L, Wang P, Ke Y, et al. Comparative transcriptome analysis between inbred lines and hybrids provides molecular insights into K+ content heterosis of tobacco (Nicotiana tabacum L). Front Plant Sci. 2022;13:940787.
Zhao J, Zhao L, Zhang M, Syed Adeel, Zafar, Fang J, et al. Arabidopsis E3 ubiquitin ligases PUB22 and PUB23 negatively regulate drought tolerance by targeting ABA receptor PYL9 for degradation. Int J Mol Sci. 2017;18:1841.
Chen T, Li Y, **e L, Hao X, Liu H, Qin W, et al. AaWRKY17, a positive regulator of artemisinin biosynthesis, is involved in resistance to Pseudomonas syringae in Artemisia annua. Hortic Res. 2021;8:217.
Zeng D, Hou P, **ao F, Liu Y. Overexpressing a novel RING-H2 finger protein gene, OsRHP1, enhances drought and salt tolerance in rice (Oryza sativa L). J Plant Biology. 2014;57:357–65.
Li H, Jiang H, Bu Q, Zhao Q, Sun J, **e Q, et al. The Arabidopsis RING finger E3 ligase RHA2b acts additively with RHA2a in regulating abscisic acid signaling and drought response. Plant Physiol. 2011;156:550–63.
Ma C, Burd S, Lers A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015;84:169–87.
Stroher E, Wang X, Roloff N, Klein P, Husemann A, Dietz KJ. Redox-dependent regulation of the stress-induced zinc-finger protein SAP12 in Arabidopsis thaliana. Mol Plant. 2009;2(002):357–67.
Acknowledgements
We thank the high-performance computing platform of Bioinformatics Center, Nan**g Agricultural University, for providing data analysis platform services.
Funding
This work was financially supported by the National Key Research and Development Program of China (2022YFD1200500), the National Natural Science Foundation of China (31870306, 32102421), the “JBGS” Project of Seed Industry Revitalization in Jiangsu Province (JBGS[2021]020), Natural Science Foundation of Jiangsu Province (BK20210395), China Agriculture Research System (CARS-23-A18) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institution.
Author information
Authors and Affiliations
Contributions
F.Z. and F.C. conceived and designed the project. L.Z., J.S., Y.N.Y. and X.Z. conducted the experiments. L.Z., J.S., and Y.Y. performed the data analysis and wrote the manuscript. J.S. and F.Z. revised the manuscript. F.Z., F.C., W.F., and Z.G. guided the research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Field and laboratory studies were conducted by local legislation. This article does not contain any studies with human participants or animals and does not involve any endangered or protected species. The plant materials sampled and experiments performed in this research complied with institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.




Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Su, J., Zhao, L., Yang, Y. et al. Comparative transcriptome analysis provides molecular insights into heterosis of waterlogging tolerance in Chrysanthemum indicum. BMC Plant Biol 24, 259 (2024). https://doi.org/10.1186/s12870-024-04954-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-04954-4