
Abstract
Milling quality (MQ) and grain shape (GS) of rice (Oryza sativa L.) are correlated traits, both determine farmers’ final profit. More than one population under multiple environments may provide valuable information for breeding selection on these MQ-GS correlations. However, suitable analytical methods for reciprocal introgression lines with linkage map for this kind of correlation remains unclear. In this study, our major tasks were (1) to provide a set of reciprocal introgression lines (composed of two BC2RIL populations) suitable for map** by linkage map** using markers/bins with physical positions; (2) to test the map** effects of different methods by using MQ-GS correlation dissection as sample case; (3) to perform genetic and breeding simulation on pyramiding favorite alleles of QTLs for representative MQ-GS traits. Finally, with four analysis methods and data collected under five environments, we identified about 28.4 loci on average for MQ-GS traits. Notably, 52.3% of these loci were commonly detected by different methods and eight loci were novel. There were also nine regions harboring loci for different MQ-GS traits which may be underlying the MQ-GS correlations. Background independent (BI) loci were also found for each MQ and GS trait. All these information may provide useful resources for rice molecular breeding.
Similar content being viewed by others

Introduction
Rice (Oryza sativa L.) is a primary food crop, feeding nearly half of the world’s population. Profit for rice farmers largely depends on not only the grain yield but also the head rice rate during grain milling [1, 2]. Grain shape and milling quality are key determinants of the head rice rate. Grain shape is often characterized by grain length (GL) and grain width (GW), which are all typical complex quantitative traits [3]. Milling quality is often characterized by three component traits, namely brown rice rate (BR), milled rice rate (MR) and head rice rate (HR), which are also typical complex quantitative traits.
Compared to piles of works on grain shape, genetic studies focusing on milling quality in rice are still lagging behind. Initial results are often based on single-population under single environment [4, 5]. Later, populations derived from Oryza glaberrima [6] and rufipogon [7] were also adopted. Recently, works for GWAS by sequenced germplasm [8, 10] also provided valuable results. For complex traits, multiple populations and/or multiple environments data may be largely beneficial to genetic dissection. Genetic control of milling quality and grain shape was identified with single population under multiple seasons [11, 13]. Data of two different populations were evaluated on QTL for milling quality and other traits [14].
Trait correlations or genetic overlaps are important especially for breeding selection. Recently, correlations between milling quality and grain yield were promoted [17,18,20, 21].
In this study, we focused on QTL map** of milling quality and grain shape using a set of reciprocal introgression lines developed by advanced backcrossing (BC2RIL) and a set of recombinant inbred lines (RIL) from a same cross. Our major tasks were (1) to construct a linkage map using a RIL population whose parents were the same as the two BC2RIL populations; (2) to conduct QTL map** of the five traits in the two BC2RIL populations based on the re-constructed linkage map. Four different map** methods were also used for comparison of detected QTLs; (3) to perform genetic and breeding simulation on pyramiding favorite alleles of QTLs for two representative traits, GL and HR.
Materials and methods
Genetic materials and genoty**
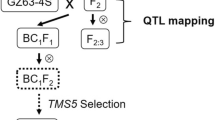
In our previous study, the development of a set of reciprocal introgression lines (BC2RILs) derived from Minghui 63 (MH63) and 02428 were described [22,23,Phenotypic data analysis Analysis of variance (ANOVA) for phenotypic data and correlation analysis among traits were performed using the AOV function in QTL IciMap** V4.2 [25]. Heritability in broad sense on the genotypic mean level in each BC2RIL population was calculated for each environment and across environments by the two equations, respectively. Best linear unbiased prediction (BLUP) values across multiple environments were calculated for each trait using the following model. where yijk was the observed phenotypic value of individual l of the genotype i in block k at environment j; μ was the mean value of the population; gi was the random effect of the ith genotype; ej was the random effect of the jth environment; r/ek(j) was the random effect of the kth block in the jth environment; geij was the random effect of the genotype by environment interaction; εijkl was the random residual effects. It’s notable that in the process of the BC2RILs construction, the recurrent parents (RPs), i.e. MH63 and 02428 were adopted as controls for phenotypic selection. According to previous experiences in reciprocal introgression line population construction [26], populations from interspecific cross in indica background tend to be more variated than that in japonica background and not easy to stabilize. Also, the phenotypic selection based on comparing to RP will largely affect the donor allelic frequency. Thus, we picked more lines with similar plant type and flowering time as MH63 during the selection of MH63-ILs, but selected more 02428-ILs largely varied from 02428 in agronomic traits except for the flowering time. Since this kind of artificial selection was conducted during the development of both BC2RIL populations, the RIL population was used for linkage map construction. Chromosome number and order of markers were anchored according to the physical map. MAP function in QTL IciMap** was adopted for estimating the genetic distance between markers. Recombination frequency was converted into map distance by the Kosambi map** function. R package LinkageMapView was used for visualization of the linkage map [27]. The algorithm of inclusive composite interval map** (ICIM) for the BC2RIL population implemented by the BIP function in QTL IciMap** [28] was used for QTL map** of the five traits in different environments, i.e. GL, GW, BR, MR and HR. QTL map** was conducted in each environment separately, as well as the BLUP values across environments. The REG method proposed by Alamin et al. [29] was also adopted for QTL map** in each environment as a comparison, which only output position and LOD score of each scanning position. As the two populations can also be viewed as CSSLs, we also used the RSTEP-LRT-ADD method in the CSL function of QTL IciMap** [30], which conducted QTL map** in each environment separately as well as the means across environments. ICIM algorithm in MET function was adopted for QTL by [31]. The LOD threshold was set at 2.5 for all the first three methods, and at 4 for MET as all environments were analyzed at the same time in MET. The scanning step was 0.5 cM. The two probabilities for entering and removing variables were set at 0.001 and 0.002, respectively. Comparison of QTL map** results among different environments, among the four map** methods, and among the five traits was then conducted. QTLs in different populations were considered to be common if the genetic positions were close enough. In other words, distance in linkage map was less than 20 cM in terms of QTL positions. In individual populations, QTLs were considered to be stable if they were identified in at least two the environments for at least one population. Twenty cM was also adopted as the minimum distance to identify pleiotropy QTLs among traits. Locus detected throughout two populations (genetic backgrounds) was important and regarded as background independent locus (BI locus). A tool named shinyCircos was used for the visualization of QTL positions on the linkage map [32]. Blib is a simulation platform for modelling, simulating, and predicting the genetic and breeding processes of different diploid species [33]. Our target of breeding design was to improve HR and GL. QTL for the two traits detected by all the four methods were considered, and those QTLs exhibiting consistent effect directions across environments were kept. For pleiotropy QTLs, if their effects for the two traits were opposite, HR was adopted as the primary index trait for determining the target genotype. The individuals mostly close to the target genotype were selected as parents for develo** simulated progenies. Three types of crosses were considered, i.e. single cross, top cross and double cross. For each cross, 1000 DH lines were generated by Blib, and proportion of target genotypes in these lines was counted.Linkage map construction using the RIL population
QTL map** for grain shape and milling quality traits in the two BC2RIL populations
Breeding simulation on pyramiding favorite alleles
Results
Phenotypic evaluation
The descriptive statistics of grain shape and milling quality traits, i.e., GL, GW, BR, MR and HR of the two BC2RIL populations in different environments were shown in Table S1. The average values of the two parents, i.e., MH63 and 02428, in each environment were from 9.12 to 10.02 and 6.38 to 7.27 cm for GL, 2.59 to 2.98 and 3.30 to 3.56 cm for GW, 75.02 to 79.91 and 73.13 to 80.88% for BR, 64.06 to 69.01 and 57.68 to 68.48% for MR, 36.94 to 57.94 and 29.59 to 64.21% for HR. The average values of progenies in populations MH63-ILs and 02428-ILs in each environment were from 6.71 to 9.93 and 6.86 to 7.84 cm for GL, 2.41 to 3.02 and 3.20 to 3.48 cm for GW, 72.06 to 78.90% and 75.83 to 79.85% for BR, 62.01 to 72.62 and 61.27 to 67.67% for MR, 35.41 to 54.41 and 47.55 to 60.35% for HR. Obvious variations were observed in phenotypic data of parents and progenies in both populations, and so was the heritability among different environments (Table S1).
Table 1 shows the variance components and heritability in broad sense of the five traits across environments. Heritability of GL and GW was considerably high, i.e., over 0.90, and heritability of BR, MR and HR was a little lower, i.e., around 0.50 to 0.71. Pearson’s correlation coefficient between the traits across environments is shown in Table 2. A significant test indicated that GL was negatively correlated with GW, and BR, MR and HR were positively correlated in both populations. HR was negatively correlated with GL, and BR was positively correlated with GW. Other correlations were insignificant or inconsistent in the two genetic backgrounds.
Linkage map constructed in the RIL population
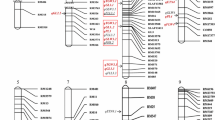
The constructed linkage map is shown in Fig. 1, and general information on the linkage map is provided in Table 3. The whole genome spanned 1572.31 cM in length, consisting of 12 chromosomes. The number of markers was 4833 and the number of unique map positions (denoted as bin) was 2448. Chromosome 5 was the longest one with the length of 200.12 cM, while chromosome 9 was the shortest with the length at 90.50 cM. The largest gap was also observed on chromosome 5 with the length at 18.44 cM. Chromosome 3 had the largest number of markers, i.e., 319 markers, and chromosome 8 had the smallest number of markers, i.e. 153 markers. The average distance between markers was 0.33 cM in the whole genome, and the average distance between bins was 0.65 cM.

Linkage map constructed from the RIL population
QTL detected by BIP
A total of 40, 28, 28, 21, and 22 QTLs were detected by BIP for GL, GW, BR, MR and HR, respectively (Table S2). QTLs detected in at least two environments for at least one population, were given in Table 4. For GL, 30 QTLs were stable and seven were detected in two genetic backgrounds (background independent, BI), i.e. qGL.BIP-2.1, qGL.BIP-3.4, qGL.BIP-3.5, qGL.BIP-3.6, qGL.BIP-4.4, qGL.BIP-4.5 and qGL.BIP-10.2. For GW, 21 QTLs were stable, two of which were BI loci, i.e. qGW.BIP-1.2 and qGW.BIP-5.1. For BR, 13 QTLs were stable, and no BI locus was found. For MR, 11 QTLs were stable, one was BI locus, i.e. qMR.BIP-2.2. For HR, 9 QTLs were stable, one BI locus, i.e. qHR.BIP-2.1 was found. Most stable QTLs had the same additive effect directions across populations and environments. For example, the additive effect of qGL.BIP-2.1 ranged from 0.09 to 0.16 in MH63-ILs, and from 0.14 to 0.18 in 02428-ILs. Some exceptions were also observed, for example, qHR.BIP-7.1 was stable in MH63-ILs, whose additive effect ranged from -2.51 to 2.02.
QTL detected by REG
A total of 35, 32, 42, 43, and 37 QTLs were detected by REG for GL, GW, BR, MR and HR, respectively, and the related information was listed in Table S3. QTLs detected in at least two environments for at least one population, were given in Table 5. For GL, 26 QTLs were stable, 7 of which were BI loci, i.e. qGL.REG-2.1, qGL.REG-2.2, qGL.REG-3.2, qGL.REG-6.1, qGL.REG-7.1, qGL.REG-9.1 and qGL.REG-12.1. For GW, 8 of 11 stable QTLs were BI loci, i.e. qGW.REG-1.2, qGW.REG-4.3, qGW.REG-5, qGW.REG-9.1, qGW.REG-9.2, qGW.REG-10.2, qGW.REG-11.2 and qGW.REG-12.3. For BR, 8 of 25 stable QTLs were BI loci, i.e. qBR.REG-2.3, qBR.REG-3.3, qBR.REG-4.1, qBR.REG-5.3, qBR.REG-7.3, qBR.REG-9.2, qBR.REG-11.2 and qBR.REG-12.3. For MR, 12 of 22 stable QTLs were BI loci, i.e. qMR.REG-1.3, qMR.REG-2.2, qMR.REG-2.4, qMR.REG-4.4, qMR.REG-5.4, qMR.REG-6.2, qMR.REG-7.1, qMR.REG-8.1, qMR.REG-8.2, qMR.REG-10.1, qMR.REG-11.1 and qMR.REG-11.3. For HR, 8 of 25 stable QTLs were BI loci, i.e. qHR.REG-1.3, qHR.REG-2.1, qHR.REG-5.3, qHR.REG-6.1, qHR.REG-6.3, qHR.REG-7.1, qHR.REG-8.1 and qHR.REG-11.3. REG detected more QTLs and more stable QTLs than did BIP. Additive effects and PVE of QTLs were not provided by the REG method.
QTL detected by CSL
A total of 36, 24, 21, 19, and 24 QTLs were detected by CSL for GL, GW, BR, MR and HR, respectively, whose information was listed in Table S4. Positions, LOD scores, PVE, and additive effect of stable QTLs were given in Table 6. For GL, 22 QTLs were stable, 8 of which were BI loci, i.e. qGL.CSL-1.1, qGL.CSL-2.1, qGL.CSL-2.2, qGL.CSL-3.4, qGL.CSL-3.5, qGL.CSL-3.6, qGL.CSL-4.2 and qGL.CSL-10.2. For GW, 13 QTLs were stable, 2 of which were BI loci, i.e. qGW.CSL-1.2 and qGW.CSL-5.1. For BR, MR, and HR, there were 8, 7, and 6 QTLs were stable, but no BI locus was found. Most stable QTLs also had the same directions of additive effect across populations and environments.
QTL detected by MET
A total of 38, 29, 25, 18, and 22 QTLs were detected by MET for GL, GW, BR, MR and HR, respectively, whose information was listed in Table S5. Since these QTLs were detected by QTL by environment interaction analysis instead of by single-environmental analysis, it is not suitable to determine if they were stably detected or not. To simplify the comparison of map** results with the other two methods, we also regarded these QTLs as stable QTLs in this study. For GL, 14 QTLs were BI loci, i.e. qGL.MET-1.1, qGL.MET-2.1, qGL.MET-2.2, qGL.MET-3.4, qGL.MET-3.5, qGL.MET-3.6, qGL.MET-4.3, qGL.MET-4.4, qGLMET-5.1, qGL.MET-6.1, qGL.MET-8.1, qGL.MET-10.1, qGL.MET-10.2, and qGL.MET-11.2. For GW, eight BI loci were detected, i.e. qGW.MET-1.2, qGW.MET-2.2, qGWMET-3.3, qGW.MET-5.1, qGW.MET-8, qGW.MET-9.1, qGW.MET-9.2, and qGW.MET-11.1. For BR, one BI locus was detected, i.e. qBR.MET-3.2. For MR, one BI locus was detected, i.e. qMR.MET-12. For HR, two BI loci were detected, i.e. qHR.MET-2.3 and qHR.MET-3.2. Positions of stable QTLs along the whole genome were shown in Fig. 2.

Stable QTLs for grain length (GL), grain width (GW), brown rice rate (BR), milled rice rate (MR), and head rice rate (HR) along the whole genome
Common loci among different QTL map** methods
All detected QTLs were used for comparison among different QTL map** methods. The number of common loci from BIP, REG, CSL, and MET were shown in Fig. 3 for all the five traits. For GL, BIP, REG, CSL, and MET detected 40, 35, 29 and 28 QTLs, respectively, of which 22 QTLs were common for the four methods. For GW, REG, BIP, CSL, and MET detected 28, 32, 25 and 29 QTLs, respectively, of which 16 QTLs were common for the four methods. For BR, BIP, REG, CSL, and MET detected 28, 42, 21 and 25 QTLs, respectively, of which 13 QTLs were common for the four methods. For MR, BIP, REG, CSL, and MET detected 21, 43, 19 and 18 QTLs, respectively, of which 13 QTLs were common for the four methods. For HR, BIP, REG, CSL, and MET detected 22, 37, 23 and 22 QTLs, respectively, among which 11 QTLs were common for the four methods. More than half of the QTLs from BIP, REG, CSL, and MET were detected by at least two map** methods, which reflected the reliability of these QTLs. The ratios of common loci (number of common loci / QTLs detected by certain method) were more than 75% by MET and CSL methods for GL, while these values decreased to 50% for HR by BIP and MET. REG detected relatively more QTLs than the other methods.

Common QTLs among different QTL map** methods
Common region harboring QTLs for different traits
Considering the trait correlations between GL and GW, and among BR, MR and HR, comparison of QTLs was conducted in two groups of traits separately, i.e. GS and MQ traits. Common regions harboring QTLs for GS and MQ traits were shown in Fig. 4 for the three map** methods separately. For GL and GW, there were 23, 19, 21 and 24 common QTLs from BIP, REG, CSL and MET, respectively. Generally, directions of additive effects of these common QTLs were opposite on the two traits. For example, qGL.BIP-3.2 and qGW.BIP-3.1 located at 30–34 cM and at 30.5 cM on chromosome 3, respectively. Additive effect of qGL.BIP-3.2 from 0.18 to 0.29 cm, while that of qGW.BIP-3.1 ranged from -0.07 to -0.04 cm. This opposite effect was consistent with the negative trait correlation between GL and GW. Similar consistencies were found for qGL.BIP-4.5 and qGW.BIP-4.3, qGL.BIP-5.1 and qGW.BIP-5.1, qGL.CSL-3.7 and qGW.CSL-3.3, etc. Some exceptional cases were also observed. For example, qGL.CSL-5.2 and qGW.CSL-5.3 at 173.73 cM and at 176.81–184.31 cM on chromosome 5, contributed additive effect from 0.14 to 0.20 and 0.04 to 0.07, respectively (Tables S2 and S4). For BR, MR and HR, there were 8, 29, 7 and 6 common QTLs from BIP, REG, CSL and MET, respectively. Although significant positive correlations were observed between the three traits, there were only several common QTLs exhibiting same effect direction of additive for all the three traits at the same time. qBR.CSL-1.1, qMR.CSL-1.1 and qHR.CSL-1.1 were located closely, and additive effects were all positive. Similarly, qBR.CSL-3, qMR.CSL-3.1 and qHR.CSL-3.1; qBR.CSL-5.2, qMR.CSL-5.2 and qHR.CSL-5.3. qBR.CSL-2.3, qMR.CSL-2.2 and qHR.CSL-2.2 were located closely, and the additive effects of them were all negative (Table S4).

Common QTLs between grain shape traits and among milling quality traits
As we mentioned, the correlation between GL and HR was also significant, and so was the correlation between GW and BR. Common QTLs between GL and HR and those between GW and BR are shown in Fig. 5. There were 17, 26, 19 and 17 common QTLs between GL and HR from BIP, REG, CSL and MET, respectively. And there were 20, 29, 11 and 17 common QTLs between GW and BR from BIP, REG, CSL and MET, respectively. It can be concluded that higher correlation between traits leaded to more common QTLs for different traits, no matter which map** method was used. Common QTLs were not only observed between traits belong to the same group, but also between traits from different groups.

Common QTLs between grain length (GL) and head rice rate (HR), and between grain width (GW) and brown rice rate (BR)
Design of the target genotype based on the identified QTLs
Nineteen QTLs for GL and HR were detected by all the four methods and had consistent directions of additive effect across different environments. Eleven of them affected both GL and HR, five of which increased or decreased both traits, and the other five had different directions of effects for the two traits. For QTLs only affecting GL, the alleles increasing GL were set as the favorable one; for QTLs only affecting HR, the alleles increasing HR were set as the favorable one; for QTLs affecting both GL and HR, the alleles increasing HR were set as the favorable one. The target genotype at the 19 QTLs is given in Table 7. Four individuals from the two populations were picked up as parents in breeding design, i.e. MH63-ILs-166, 02428-ILs-185, MH63-ILs-65 and MH63-ILs-168, which had only 4, 5, 5 and 5 different QTL genotypes compared with the target genotype. Their genotypes were also given in Table 7. The predicted values of the selected lines with these QTLs under certain environments were also provided (Table S6).
For the four selected individuals, we performed two single crosses, six top crosses and six double crosses to generate the target genotype, respectively, by using Blib. These crosses and the corresponding percentage of the designed genotype in the DH population were also provided in Table 8. The percentage of the designed genotype for SC1 was 0.0014, higher than that for SC2. Percentage for TC2 was 0.0022, which was the highest among the six top crosses. Percentage for DC2 was 0.0018, which was the highest among the six double crosses. TC2 and DC2 had the common three parents, but different cross design led to different genetic structures in the two populations, as well as different percentages of the designed genotype.
Discussion
More reasonable map** results based on linkage map
In previous map** work with BC2RIL [23, 25], linkage map would be preferred. We compared our map** results based on new map with same map** method of BIP (Table S2) to our previous report [2. For QTLs detected by BIP and CSL, reliability of these QTLs was higher than those stable in only one population, and followed by the unstable QTLs. As we presented before, directions of additive effects of these QTLs were mostly consistent for the two populations. However, values of additive effects varied significantly across populations and environments. Generally, if a QTL for the two populations was detected in the same environment, similar or larger effects was observed for MH63-ILs than that for 02428-ILs. For example, qGL.BIP-10.2 was detected in six environments for MH63-ILs and five environments for 02428-ILs (Table 4 and S2). In environments SY, SZ, XZ, and BLUP, qGL.BIP-10.2 was detected for both populations. Its additive effect was larger in MH63-ILs than that in 02428-ILs except in SZ. The larger QTL effects may be related to the stricter selection on MH63-ILs. Further study is needed to prove this inference.
QTLs regions with multiple effects
As we mentioned above, some common QTLs were detected within and also between GS and MQ trait groups. As an example, a total of nine regions harboring these QTLs are located within an interval shorter than 20 cM (Table S7). Of these regions, three were located on chromosome 5, with an average size of 17.9 cM corresponding to about 1.6 Mb; two on chromosome 3 with an average size of 3.8 cM (2.4 Mb); other four with averaged size of 4.2 cM (0.5 Mb) on chromosomes 1, 2, 10, and 11 respectively. Especially, qGL.BIP-3.4 which contains both the major effect QTL for grain length, GS3 [Application of detected QTLs in rice breeding A large amount of QTL map** studies have been conducted for various traits in rice in the last two decades, and how to utilize so many detected QTLs or genes becomes a challenge to the breeders. In this study, the simulation platform Blib has been adopted to design the target genotypes using identified QTLs. Two significantly related traits, GL and HR, were selected as examples to demonstrate the design process, which was quite important in plant breeding. According to the simulation results, the target genotype with larger HR and relatively longer GL could be obtained when 1000 DHs were generated from the cross between or among the selected individuals in the two map** populations, no matter single cross, top cross, or double cross was adopted. The two map** populations in this study are generated by consecutive backcrossing, which are close to CSSL populations in two directions, the MH63-ILs using MH63 as the recurrent parent, and the 02428-ILs using 02428 as the recurrent parent. The individuals of MH63-ILs had higher genotypic similarity with the parent MH63, of which 91.30% were same as parent MH63, 7.08% were same as parent 02428, and 1.62% were missing. Genotypes of individuals in 02428-ILs were closer to parent 02428, among which 19.88% was same as parent MH63, 76.55% was the same as parent 02428, and 3.57% was missing. But more abundant genetic variations can be found in the two populations than the two parents. When the target genotype is defined in simulation study, it is easy to select suitable parents for crossing, which are in genotype at limited number of QTL. In addition, these parents are complementary, i.e. at least one of the parents was homogenous with target genotype. The development of map** populations and simulation tools and procedures for the target genotype provides a potential way of utilizing of the detected QTLs.
Conclusions
A set of reciprocal introgression lines with a genetic map was provided for genetic dissection of complex traits. The procedure of selecting more suitable analytical methods for this kind of population were also presented by using MQ-GS trait correlations / genetic overlaps as example. Besides eight new loci, nine loci-harboring regions for different traits and background independent loci were reported. Background independent (BI) loci were also found for each MQ and GS traits. All these information together with the simulation on breeding design may provide useful guidelines for rice molecular breeding.
Availability of data and materials
Data for the 424 BC2RIL lines including the genotypic data, maps and seed availability are provided through the following URL https://rfgb.rmbreeding.cn/download/publicDataDownload. Seeds can be requested through the following URL https://rfgb.rmbreeding.cn/search/germplasm/seedrequest or email to the corresponding authors.
Abbreviations
- MQ:
-
Milling quality
- GS:
-
Grain shape
- RIL:
-
Recombinant inbred line
- GL:
-
Grain length
- GW:
-
Grain width
- FGB:
-
Functional Genomic Breeding
- BR:
-
Brown rice rate
- MR:
-
Milled rice rate
- HR:
-
Head rice rate
- QTL:
-
Quantitative trait locus
References
Butardo VM Jr, Sreenivasulu N. Improving head rice yield and milling quality: State-of-the-Art and future prospects. In: Rice grain quality-methods and protocols. vol. 1892. 2019. p. 1–18.
Bao J. 10 - Rice milling quality. In: Rice (Fourth Edition). Edited by Bao J: AACC International Press; 2019. p. 339–369. https://www.sciencedirect.com/book/9780128115084/rice#book-description.
Yin C, Li H, Li S, Xu L, Zhao Z, Wang J. Genetic dissection on rice grain shape by the two-dimensional image analysis in one japonica × indica population consisting of recombinant inbred lines. Theor Appl Genet. 2015;128(10):1969–86.
Tan YF, Sun M, ** quantitative trait loci for milling quality, protein content and color characteristics of rice using a recombinant inbred line population derived from an elite rice hybrid. Theor Appl Genet. 2001;103:1037–45.
Mei HW, Luo LJ, Guo LB, Wang YP, Yu XQ, Ying CS, Li ZK. Molecular map** of QTLs for rice milling yield traits. Acta Genet Sin. 2002;29(9):791–7.
Aluko G, Martinez C, Tohme J, Castano C, Bergman C, Oard JH. QTL map** of grain quality traits from the interspecific cross Oryza sativa x O. glaberrima. Theor Appl Genet. 2004;109(3):630–9.
Liu J, Kui L, Zhu Z, Tan L, Wang G, Li Q, Shu J, Sun C. Identification of QTLs associated with processing quality and appearance quality of common wild rice (Oryza rufipogon Griff.). J Agric Biotechnol. 2007;15(1):90–6.
Wang X, Pang Y, Wang C, Chen K, Zhu Y, Shen C, Ali J, Xu J, Li Z. New candidate genes affecting rice grain appearance and milling quality detected by genome-wide and gene-based association analyses. Front Plant Sci. 1998;2017:7.
Qiu X, Pang Y, Yuan Z, **ng D, Xu J, Dingkuhn M, Li Z, Ye G. Genome-wide association study of grain appearance and milling quality in a worldwide collection of indica rice germplasm. PLoS One. 2015;10(12):e0145577.
Ren D, Rao Y, Huang L, Leng Y, Hu J, Lu M, Zhang G, Zhu L, Gao Z, Dong G, et al. Fine Map** Identifies a New QTL for Brown Rice Rate in Rice (Oryza Sativa L.). Rice. 2016;9(1):4.
Mei DY, Zhu YJ, Fan YY. Map** QTL for rice milling and appearance quality traits in indica rice. Yi Chuan. 2012;34(12):1591–8.
Luo LL, Zhang YX, Chen DB, Zhan XD, Shen XH, Cheng SH, Cao LY. QTL map** for hull thickness and related traits in hybrid rice **eyou 9308. Rice Sci. 2014;21(1):29–38.
Nelson JC, McClung AM, Fjellstrom RG, Moldenhauer KA, Boza E, Jodari F, Oard JH, Linscombe S, Scheffler BE, Yeater KM. Map** QTL main and interaction influences on milling quality in elite US rice germplasm. Theor Appl Genet. 2011;122(2):291–309.
Swamy BPM, Kaladhar K, Shobha Rani N, Prasad GSV, Viraktamath BC, Reddy GA, Sarla N. QTL analysis for grain quality traits in 2 BC2F2 populations derived from crosses between Oryza sativa cv Swarna and 2 accessions of O. nivara. J Hered. 2012;103(3):442–52.
Zhang H, Zhu Y-J, Zhu A-D, Fan Y-Y, Huang T-X, Zhang J-F, **e H-A, Zhuang J-Y. Identification and verification of quantitative trait loci affecting milling yield of rice. Agronomy. 2020;10(1):75.
Xu ZJ, Chen WF, Ma DR, Lu YN, Zhou SQ, Liu LX. Correlations between rice grain shapes and main qualitative characteristics. Acta Agron Sin. 2004;30(9):894–900.
Zheng TQ, Xu JL, Li ZK, Zhai HQ, Wan JM. Genomic regions associated with milling quality and grain shape identified in a set of random introgression lines of rice (Oryza sativa L.). Plant Breeding. 2007;126(2):158–63.
Hu X, Shi Y, Jia Q, Xu Q, Wang Y, Chen K, Sun Y, Zhu L, Xu J, Li Z. Analyses of QTLs for rice panicle and milling quality traits and their interaction with environment. Acta Agron Sin. 2011;37(07):1175–85.
Li ZF, Wan JM, **a JF, Zhai HQ, Ikehashi H. Identification of quantitative trait loci underlying milling quality of rice (Oryza sativa) grains. Plant Breeding. 2004;123(3):229–34.
Wang J, Wan X, Li H, Pfeiffer WH, Crouch J, Wan J. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor Appl Genet. 2007;115(1):87–100.
Wang J, Chapman SC, Bonnett DG, Rebetzke GJ, Crouch J. Application of population genetic theory and simulation models to efficiently pyramid multiple genes via marker-assisted selection. Crop Sci. 2007;47(2):582–8.
Nafisah. QTL dissection of source and sink related traits using reciprocal introgression lines in rice (Oryza sativa L.) under different water regimes. Graduate School, Chinese Academy of Agricultural Sciences; 2013.
Zhang Q, Zheng T-Q, Hoang L, Wang C-C. Nafisah, Joseph C, Zhang W-Z, Xu J-L, Li Z-K: Joint map** and allele mining of the rolled leaf trait in rice (Oryza sativa L.). PloS one. 2016;11(7):e0158246.
Qiu X, Chen K, Lv W, Ou X, Zhu Y, **ng D, Yang L, Fan F, Yang J, Xu J, et al. Examining two sets of introgression lines reveals background-independent and stably expressed QTL that improve grain appearance quality in rice (Oryza sativa L.). Theor Appl Genet. 2017;130(5):951–67.
Meng L, Li H, Zhang L, Wang J. QTL IciMap**: Integrated software for genetic linkage map construction and quantitative trait locus map** in biparental populations. The Crop J. 2015;3(3):269–83.
Mei HW. Molecular Map** of Important Agronomic Traits in Rice using Reciprocal Introgression Line (IL) Populations. Wuhan: Huazhong Agricultural University; 2005.
Ouellette LA, Reid RW, Blanchard SG, Brouwer CR. LinkageMapView—rendering high-resolution linkage and QTL maps. Bioinformatics. 2017;34(2):306–7.
Li HH, Ye GY, Wang JK. A modified algorithm for the improvement of composite interval map**. Genetics. 2007;175(1):361–74.
Alamin M, Sultana MH, Xu H, Mollah MNH. Robustification of linear regression and its application in genome-wide association studies. Front Genet. 2020;11:549.
Wang J, Wan X, Crossa J, Crouch J, Weng J, Zhai H, Wan J. QTL map** of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet Res. 2006;88:93–104.
Li S, Wang J, Zhang L. Inclusive composite interval map** of QTL by environment interactions in biparental populations. PLoS ONE. 2015;10(7):e0132414.
Yu Y, Ouyang Y, Yao W. shinyCircos: an R/Shiny application for interactive creation of Circos plot. Bioinformatics. 2018;34(7):1229–31.
Zhang L, Li H, Wang J. Blib is a multi-module simulation platform for genetics studies and intelligent breeding. Commun Biol. 2022;5(1):1167.
Wang CC, Yu H, Huang J, Wang WS, Faruquee M, Zhang F, Zhao XQ, Fu BY, Chen K, Zhang HL, et al. Towards a deeper haplotype mining of complex traits in rice with RFGB v.20. Plant Biotechnol J. 2020;18(1):14–6.
Zhang G-M, Zheng T-Q, Chen Z, Wang Y-L, Wang Y, Shi Y-M, Wang C-C, Zhang L-Y, Ma J-T, Deng L-W, et al. Joint Exploration of Favorable Haplotypes for Mineral Concentrations in Milled Grains of Rice (Oryza sativa L.). Front Plant Sci. 2018;9:447.
Fan C, **ng Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet. 2006;112:1164–71.
Funding
This work was mainly supported by the National Key Research and Development Program of China (2020YFE0202300), National Nature Science Fund of China (32171993), Key Special Program (2022ZD0400404), Ministry of Science and Technology, China, International Science & Technology Innovation Program of Chinese Academy of Agricultural Sciences (CAASTIPS, CAAS-ZDRW202109), Guangxi Key Laboratory of Rice Genetics and Breeding (2022–36-Z01-KF10), Key Research and Development Program of Hainan (ZDYF2021XDNY128), and the Bill & Melinda Gates Foundation (OPP1130530).
Author information
Authors and Affiliations
Contributions
Tianqing Zheng and Jianlong Xu designed and conceived research; Mwenda Emelin, **an** Qiu, Fangjun Fan, Luyan Zhang and Md. Alamin performed data collection and analysis; Muhiuddin Faruquee, Hui Hu, Junying Xu, Jie Yang, Haiming Xu, Jauhar Ali, Bailong Liu, Yumin Shi, and Zhikang Li give valuable suggestions and contributed new analytic tools; Luyan Zhang, Tianqing Zheng, and Jianlong Xu draft the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Summary statistics of the five traits for the two populations in different environments. Table S2. Detail information of all QTLs detected by BIP. Table S3. Detail information of all QTLs detected by REG. Table S4. Detail information of all QTLs detected by CSL. Table S5. Detail information of all QTLs detected by MET. Table S6. Predicted genotypic values of the selected lines and target genotype in each population and environment. Additive effect of each QTL for calculating the genotypic values was picked up from the BIP map** results at the nearest position of the average QTL position across in Table 7. Table S7. Nine regions harboring QTLs for MQ-GS traits.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Emelin, M., Qiu, X., Fan, F. et al. Identification of reliable QTLs and designed QTL breeding for grain shape and milling quality in the reciprocal introgression lines in rice. BMC Plant Biol 24, 38 (2024). https://doi.org/10.1186/s12870-023-04707-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04707-9