
Abstract
Background
The architecture of inflorescence in crops is a key agronomic feature determining grain yield and thus has been a major target trait of cereal domestication.
Results
In this study, we show that a simple spreading panicle change in rice panicle shape, controlled by the Spreading Panicle 9 (SPR9) locus, also has a significant impact on the resistance to rice false smut (RFS). Meanwhile, we mapped a novel spr9 mutant gene between markers Indel5-18 and Indel5-22 encompassing a genomic region of 43-kb with six candidate genes. Through gene prediction and cDNA sequencing, we confirmed that LOC_Os05g38520 is the target gene in the spr9 mutant, which encodes 60 S ribosomal protein L36-2. Further analysis showed that the spr9 mutant is caused by a 1 bp deletion in the first exon that resulted in premature termination. Knockout experiments showed that the SPR9 gene is responsible for the spreading panicle phenotype of the spr9 mutant. Interestingly, the spr9 mutant was found to improve resistance to RFS without affecting major agronomic traits. Taken together, our results revealed that the spr9 allele has good application prospects in rice breeding for disease resistance and panicle improvement.
Conclusions
We report the map-based cloning and functional characterization of SPR9, which encodes a 60 S ribosomal protein that regulates spreading panicles and affects the resistance to false smut in rice.
Similar content being viewed by others


Background
Plant inflorescence configuration is a key agronomic factor affecting mine grain yield and a major target for crop domestication and improvement [1]. Understanding the genetic basis of crop inflorescence structure will not only help to clarify the evolutionary mechanism of crops but also help to improve crop yield.
Rice (Oryza sativa. L) is one of the most important food crops in the world, and its panicle morphological development and molecular regulation mechanism have been the focus of research. In recent years, some progresses have been made in the study of panicle development of rice, a monocot model plant, but it is far less detailed than that Arabidopsis thaliana [2]. The panicle formation process of rice is a complex physiological and biochemical process involving axillary meristem development, inflorescence structure building and grain development. The in-depth study of panicle formation will not only help to reveal the regulatory mechanism of panicle morphogenesis but also provide theoretical guidance for the improvement of panicle type in rice [3]. Genes related to rice panicles, such as Gn1a/OsCKX2 [4], DEP1 [24], spr4 [25], spr5 [26], spr8 [27] and OsLG1 [1]. However, of these localized genes or QTLs, only OsLG1 was successfully cloned as the SPR3 locus [24]. Further studies demonstrated that a single nucleotide polymorpho-6 (SNP6) in the 11 kb cis-regulatory region upstream of the transcriptional start site of OsLG1 gene resulted in a compact panicle type in cultivated rice during rice domestication, but it did not change its expression in the leaf tongue, resulting in a compact panicle type and normal leaf tongue development in cultivated rice [1, Development of molecular markers Insertion-deletion (InDel) markers were designed by manual comparison of genome sequences between japonica (cv. Nipponpare) and indica (cv. 93 − 11), and primers were designed to map the polymorphic regions of rice subspecies using Primer Premier 5.0. Plant DNA and DNA amplification was performed by polymerase chain reaction (PCR) with minor modifications [36]. The PCR products were separated by 8% polyacrylamide denaturing gel electrophoresis, and the molecular markers were stained with silver [37]. Markers of target genes were identified by bulked segregant analysis. Leaf DNA of 20 mutant plants randomly selected from the F2 population were used for construction of a mutant DNA library. SSR markers distributed in the rice genome were used to amplify the spr9 mutant DNA, and Hui1586 DNA was used as a control for linkage detection. The marker band of the mutant gene was the same as the marker band of the spr9 mutant. The band types of the mutant (spr9 spr9) and wild type (SPR9 SPR9) were denoted as 1 and 3, respectively; 2 was used to represent a heterozygote (spr9 SPR9). Linkage analysis was performed using MAPMAKER version 3.0 software [38], and the linkage map was basically the same as the linkage map reported previously [39]. First, 326 SSR markers were screened from the rice molecular map to study the polymorphisms of spr9 and Hui1586. The results indicated that 253 pairs of primers showed polymorphism. Twenty plants with mutant phenotype and 20 plants like wild type selected from the F2 population, respectively, and linkage analysis of the spr9 locus was performed using these 253 polymorphic markers. Second, to narrow the map** region, we identified 1452 mutants from the F2 population of spr9 × Hui1586. By comparing Nipponbare and the indica cultivar 93 − 11 (http://www.elabcaas.cn/rice/index.html), InDel markers in the open rice genome sequence were designed to predict the likelihood of polymorphism between spr9 and Hui1586. Candidate genes were predicted based on existing sequence annotation databases (http://rice.plantbiology.msu.edu/; http://www.tigr.org/). Clones were fixed on the target gene combination mark with the basic local alignment search tool (BLAST) in pairs (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) for sequence alignment. The DNA sequences of spr9 and SPR9 were used for a complete alignment with Clustal X version 1.81. The CRISPR-plant database and website were used to design gRNA spacer sequences with high specificity (Supplementary Table 1) [40]. A gRNA interval spanning the first exon of the gene was used to target the SPR9 gene of Hui1586. The transformation and identification of the edited rice lines were performed by Wuhan Boyuan Technology Company. After obtaining the edited transgenic plants, PCR products of transgenic CRISPR-edited lines were sequenced to identify specific mutations [41]. Primers for the CRISPR/Cas9 study are listed in Supplementary Table 1. A GFP-SPR9 fusion was constructed 35 S: SPR9-pSuper1300-GFP and transformed to Agrobacterium tumefaciens GV3101, which was used to infiltrate tobacco leaves, and the expression of GFP was observed by laser confocal microscopy (Zeiss 880) .PCR amplification and molecular marker detection
Bulked segregant analysis
Molecular map** of the spr9 gene
Bioinformatics analysis
CRISPR mediated editing
GFP fusion and subcellular localization
Results
Analysis of the main agronomic traits of spr9
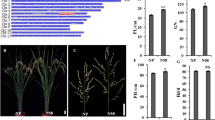
To elucidate the regulatory genes involved in spike development in rice, we screened and obtained a spr9 mutant with changes in spike traits in the R20-1 background, which displayed a spread panicle (Fig. 1a) and the corresponding wild type gene was named Spreading Panicle 9 (SPR9). Phenotype comparisons between the spr9 mutant and wild-type R20-1 are shown in Table 1. The results showed that there were no significant differences in plant height, panicle length, effective panicle number, number of grains per panicle, seed setting rate, 1000-grain weight, grain length or grain width between the spr9 mutant and the wild type (Fig. 1b-I and Supplementary Table 2).

Phenotypic comparison of the spr9 mutant and wild type R20-1. a: The spr9 mutant showed the phenotype of spreading panicles compared with R20-1. b, c, d, e, f, g, h, and i indicate no differences in plant height, panicle length, number of effective panicles, spikelets per panicle, seed setting rate, 1,000-grain weight, grain length and grain width between R20-1 and the spr9 mutant, respectively
Resistance analysis of RFS between the spr9 mutant and R20-1
A certain relationship was proposed between rice panicle type and the resistance level of RFS [30, 31]. To further compare whether there is a difference in resistance in RFS between the spr9 mutant and R20-1, we inoculated the spr9 mutant and R20-1 plants by manual injection, and each material was set up in triplicate. The experimental results showed that the disease score of the spr9 mutant was 3 and that of R20-1 was 4, further suggesting that the spr9 mutant exhibited more resistance to rice smut than the R20-1 control (Fig. 2a and b).

Disease symptoms of the spr9 mutant and R20-1 infected by U. virens. a: Rice smut resistance of the spr9 mutant and R20-1 plants using injected inoculation in a greenhouse. Three replications were carried out, each individual was artificially inoculated with U. virens in three panicles. Inoculated plants were kept in a greenhouse at 27 °C with 90–100% RH for 7 days. Then, they were placed at 27 °C and 80% RH until rice false smut symptoms appeared. b: The disease score of the spr9 mutant is 3 and that of R20-1 is 4. The spr9 mutant exhibited more resistance to rice smut than R20-1. Asterisks indicate statistical significance (p < 0.01) determined by Student’s t-test
Genetic analysis of the spr9 mutant
To determine the genetic mode of spr9 mutant, we generated two kinds of hybrid F1 plants by crossing spr9 mutant with R20-1 and R20-1 with spr9, respectively. Both kinds of F1 plants showed same panicle phenotype as the wild type, indicating that the spr9 was recessive heredity and independent of cytoplasm (Table 1). Then in the two F2 populations, we found that the panicle phenotype of plants as spr9 mutant and R20-1 wild type fits a 1:3 segregation ratio (χ2 = 0.118~0.386, P > 0.5), implying a single gene controlled the spr9 mutant phenotype. Together, these results indicate that the spr9 mutant is genetically controlled by a single recessive gene.
Preliminary molecular map** of the spr9 gene
To determine the gene underlying the spr9 mutant, we further conducted a gene map** population by crossing the spr9 mutant with the japonica rice cultivar Hui1586. Total of 253 SSR markers showed polymorphism between spr9 and Hui1586 were obtained. Among these polymorphic markers, RM8211, on chromosome 5, showed a complete co-segregation with mutant phenotype in the selected 20 F2 plants with mutant phenotype and 20 plants with wild type phenotype. Furthermore, 193 recessive plants from the F2 population were genotyped by RM8211 and RM5970, recombinant plants were found, verifying the linkage relationship between the marker and spr9 mutant phenotype. Subsequently, we found one other polymorphic marker RM5970, located on chromosome 5, also showed linkage with spr9 mutant phenotype. The recombinants identified from RM8211 all showed homozygous genotype as spr9 mutant at the RM5970 locus, and the recombinants from RM5970 presented heterozygous or wild homozygous genotype at RM8211 locus, therefore, we preliminarily mapped the spr9 gene into the region of RM8211 and RM5970. Thus, spr9 was preliminarily located in a 16.5 cM region between SSR markers RM8211 and RM5970 on chromosome 5 (Fig. 3a).

Physical maps and structural comparison of the spr9 gene. a: Primary map** of the spr9 gene. The gene was mapped to the region between the markers RM8211 and RM5970. b: Further map** of the spr9 gene. The gene was mapped to the region between commonmarkers RM1090 and RM1187. c: Accurate map** of the spr9 gene. The spr9 was mapped to the region between markers Indel15-5 and Indel15-6 selected from 10 newly developed InDel markers. d: Fine map** of the spr9 gene. The spr9 was localized to a 43-kb region between the markers Indel15-18 and Indel15-22 selected from 21 newly developed InDel markers, and the recombinant number between the markers and target genes is indicated under the linkage map. e: Candidate genes in the 43-kb target region. f: SPR9 has three exons, and spr9 exhibits a 1 bp deletion in the first exon
Fine map** of the spr9 gene
To localize the spr9 gene in a smaller region, we constructed a genetic map between RM8211 and RM5970. Two pairs of polymorphic primers RM1090 and RM1187 were screened from the common primers between RM8211 and RM5970. Furthermore, 193 recessive plants from the F2 population were genotyped by RM1090 and RM1187, recombinant plants were found, verifying the linkage relationship between the marker and spr9 mutant phenotype, spr9 was located between the RM1090 and RM1187 molecular markers, and the distance between the two molecular markers was 5.9 cM (Fig. 3b). To further localize the spr9 gene, 1452 recessive plants from the F2 population were genotyped by Indel5-3, Indel5-5, Indel5-6 and Indel5-10 (Table 2), recombinant plants were found, verifying the linkage relationship between the marker and spr9 mutant phenotype. The localization results showed that the spr9 gene was located between the molecular markers Indels 5–5 and 5–6, and the physical distance between the two markers was 477 kb (Fig. 3c; Table 2). To accurately locate the spr9 gene, 1452 recessive plants from the F2 population were also genotyped by 10 polymorphic InDel markers Indel5-12, Indel5-15, Indel5-16, Indel5-18, Indel5-20, Indel5-22, Indel5-25, Indel5-26, Indel5-29 and Indel5-31 (Table 2), recombinant plants were found, verifying the linkage relationship between the marker and spr9 mutant phenotype. The result showed 10 polymorphic InDel markers were detected 20, 9, 3, 1, 0, 1, 4, 8, 12 and 35 recombinant plants, respectively (Fig. 3d). Therefore, we accurately located the spr9 gene between the Indel5-18 and Indel5-22 molecular markers, and the physical distance between them was 43-kb (Fig. 3d).
Candidate gene analysis in the 43-kb region
The functions of six candidate genes were annotated (LOC_Os05g38500, LOC_Os05g38510, LOC_Os05g38520, LOC_Os05g38530, LOC_Os05g38540 and LOC_Os05g38550) in this 43-kb region (Fig. 3e). According to the database annotation, the results showed that each of the six candidate genes had a corresponding full-length cDNA.
To find which gene is responsible for the mutant phenotype, we sequenced the above six genes in R20-1 and the spr9 mutant, and the results showed that there was only a 1-bp deletion (T) (LOC_Os05g38520) between wild-type R20-1 and the spr9 mutant (Fig. 3f). No difference were observed in the sequences of other five genes. Therefore, we hypothesized that LOC_Os05g38520 corresponds to SPR9. Open reading fragment analysis showed that the SPR9 gene (LOC_Os05g38520) had three exons and two introns (Fig. 3f).
The spr9 gene is responsible for the spread panicle phenotype
To determine the phenotype of spr9 in the japonica genetic background, we examined whether knockout of SPR9 in the cultivar Hui1586 (japonica) would lead to the spread panicle phenotype. Using the CRISPR/Cas9 gene editing system, a sequence-specific guide RNA (sgRNA) was designed to knock out the SPR9 gene. We obtained a total of three homozygotes from three independent knockout events and confirmed their presence of insertion and deletion mutations at the target sites by Sanger DNA sequencing (Fig. 4a). We then investigated and analysed the panicle characteristics of three homozygous lines after maturity and found that all three homozygous lines showed a panicle spread phenotype (Fig. 4b), which indicated that knockout of the SPR9 gene in Hui1586 would lead to the spread panicle phenotype. In addition, analysis of other agronomic traits showed that there were no significant differences in plant height, panicle length, effective panicle number, number of grains per panicle, seed setting rate, 1000-grain weight, grain length or grain width between Hui1586 and three knockout transgenic lines (Supplementary Table 3). These results were consistent with the agronomic traits of the spr9 mutant in the R20-1(wild-type). Taking together, we concluded that spr9 gene was the causal gene for the spread panicle phenotype in the spr9 mutant.

Knockout transgenic lines showed the phenotype of the spr9 mutant. a: Three independent events (designated SPR9-KO-Line1, SPR9-KO-Line2 and SPR9-KO-Line3) were generated using the CRISPR/Cas9 system and verified by sequencing. b: Panicle differences between Hui1586 and three knockout lines. The three knockout lines generated by CRISPR/Cas9 all exhibit the phenotype of spreading panicles
Expression pattern and subcellular localizationof SPR9
To further understand the function of SPR9, reverse transcription- quantitative PCR (RT‒qPCR) was used to detect the expression patterns of SPR9 at different developmental stages of rice (Primers are shown in Supplementary Table 1). The results showed that SPR9 was expressed in all tissues tested here, including roots, shoots, and leaves of two- and four-week-old panicles of 0.5–1 cm, 1–3 cm, 3–5 cm, and 5–10 cm length, along with germinating and mature seeds and callus, but it was predominantly expressed in the seed (germination) and in panicles (5–10 cm length) (Fig. 5).

The expression patterns of SPR9. The expression patterns of SPR9. RNA samples were extracted from different tissues of Jiafuzhan, including roots, shoots, and leaves of two-, four- and six-week-old seedlings, spikelets of 0.5–1 cm, 1–3 cm, 3–5 cm, and 5–10 cm in length, germinating and mature seeds and callus. Data represent the mean and standard deviation of three biological replicates. Three technical replicates for each biological sample were used. The error bar represents the standard deviation (SD) of the value from three independent biological samples
In order to further analyze the localization of SPR9, we constructed 35 S: SPR9-pSuper1300-GFP vector (Primers are shown in Supplementary Table 1) and transformed to Agrobacterium tumefaciens GV3101. Three days after injecting tobacco leaves, the localization of SPR9 cells was observed by laser confocal microscopy (The Zeiss 880 confocal microscope). The results showed the localization of SPR9 in the nucleus (Fig. 6).

The subcellular localizationof SPR9. The localization of SPR9-pSuper1300-GFP in Nicotiana tabacum L. cells was observed by laser confocal microscopy. The results showed that SPR9-pSuper1300-GFP mainly expressed in the nucleus, Bar = 20 μm
Discussion
SPR9 is a new spread panicle-related gene
The spread panicle trait is a typical mutant trait in rice, and there have been some reports on its gene map**. To date, a total of 7 genes for spike traits have been reported, namely, spr1 [22], spr2 [23], spr3[24], spr4 [25], spr5[26], spr8 [27] and OsLG1[1]. Among these genes, spr1 and spr8 were controlled by a recessive nuclear gene, and other five sprouting trait genes were controlled by dominant genes. In this study, the spr9 gene was identified as a recessive mutation. Through further comparison, no cloned spread panicle gene was found in this region, so we speculated that spr9 was a new gene.
Why does spr9 show enhanced resistance to RFS?
SPR9 encodes the 60 S ribosomal protein L36-2, which is a ribosome associated protein. However, why does spr9 affect the disease resistance of rice? Studies have shown that posttranslational reprogramming is another fundamental regulatory pathway of plant immunity and specifically regulates translation in response to PAMP-triggered immunity induction [42]. Consistent with this result, previous transcriptomic studies of rice Pi21-silenced plants infected by Magnaporthe oryzae suggested that ribosomes were a third enrichment pathway compared to Nipponbare plants [43]. A recent study showed that translation was the most significantly enriched term in Gene Ontology (GO) analysis, while ribosomes were the most significantly enriched pathway in the Kyoto Encyclopaedia of Genes and Genomes (KEGG) analysis [46]. In rice, it was found that the spike characters were related to the occurrence of RFS. The rice varieties with vertical close ear type are more susceptible to RFS, while the rice varieties with scattered ear type and long curved ear type are not [30, 31]. We speculated that the main reason for the relatively better resistance of spread or long-curved rice panicle types is that under the same environmental conditions, spread or long-curved rice panicle types have better permeability, short duration of high humidity conditions, and relatively low humidity. More importantly, studies have shown that the best condition for rice false smut is low temperature and high humidity [47, 48]. Therefore, this is consistent with the results of this study that the spr9 mutants exhibit better disease resistance to RFS.
The application prospect of the spr9 gene in rice breeding
Many wild rice species have typical spread spikelets because the spread spikelets help them to improve the outcrossing seed setting rate and reproductive ability in the field to better adapt to the environment [49, 50]. At the same time, some studies have shown that the spread of panicle traits is often linked with some undesirable agronomic traits, such as shorter plant height, fewer tillers, fewer branches and stalks, lower yield, and stronger grain setting, but the grain quality is better[49, 50].
In this study, most importantly, our data indicated that the spr9 mutant not only enhanced resistance to RFS but also did not affect the important agronomic traits of rice (Figs. 1 and 2 and Supplementary Table 2). Together, the results indicate that SPR9 has good application prospects in future rice disease resistance and improved panicle breeding. For example, to improve the outcrossing rate of male sterile lines, we can transfer spr9 into sterile lines. At the same time, to improve rice smut resistance to a certain extent, we transferred spr9 into restorer lines.
Conclusion
In this study, a novel SPR9 gene was mapped and identified as a ribosomal protein coding gene. Then, CRISPR/Cas9 knockout experiments confirmed that the SPR9 gene is responsible for the spreading panicle phenotype of the spr9 mutant. Importantly, the spr9 mutant was found to improve resistance to RFS without affecting major agronomic traits, indicative of potential applications of spr9 in broader breeding programs. Taken together, our results revealed that the spr9 gene has good application prospects in future rice disease resistance and improved panicle breeding.
Data Availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. The genome sequence of LOC_Os05g38520 (SPR9) can be found in the NCBI database (http://www.ncbi.nlm.nih.gov/), and the number of GenBank is AK058918.
References
Ishii T, Numaguchi K, Miura K, Yoshida K, Thanh PT, Htun TM, Yamasaki M, Komeda N, Matsumoto T, Terauchi R, Ishikawa R, Ashikari M. OsLG1 regulates a closed panicle trait in domesticated rice. Nat Genet. 2013;45(4):462–5.
Huang XH, Huang SW, Han B, Li JY. The integrated genomics of crop domestication and breeding. Cell. 2022;185(15):2828–39.
Liu Q, Han RX, Wu K, Zhang JQ, Ye YF, Wang SS, Chen JF, Pan YJ, Xu XP, Zhou JW, Tao DY, Wu YJ, Fu XD. G-protein βγ subunits determine grain size through interaction with MADS-domain transcription factors in rice. Nat Commun. 2018;9(1):852.
Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angles ER, Qian Q, Kitano H, Matsuoka M. Cytokinin oxidase regulates rice grain production. Science. 2005;309(5735):741–5.
Huang XZ, Qian Q, Liu ZB, Sun HY, He SY, Luo D, **a GM, Chu CC, Li JY, Fu XD. Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet. 2009;41(4):494–7.
Huang HX, Ye YF, Song WZ, Li Q, Han RX, Wu CC, Wang SX, Yu JP, Liu XY, Fu XD, Liu Q, Wu K. Modulating the C-terminus of DEP1 synergistically enhances grain quality and yield in rice. J Genet Genomics. 2022;49(5):506–9.
Wu Y, Wang Y, Mi XF, Shan JX, Li XM, Xu JL, Lin HX. The QTL GNP1 encodes GA20ox1, which increases grain number and yield by increasing cytokinin activity in rice panicle meristems. PLoS Genet. 2016;12(10):e1006386.
Jiao YQ, Wang YH, Xue DW, Wang J, Yan MX, Liu GF, Dong GJ, Zeng DL, Lu ZF, Zhu XD, Qian Q, Li JY. Regulation of OsSPL14 by OsmiR156 defifines ideal plant architecture in rice. Nat Genet. 2010;42(6):541–4.
Miura K, Ikeda M, Matsubara A, Song XJ, Ito M, Asano KJ, Matsuoka M, Kitano H, Ashikari M. OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat Genet. 2010;42(6):545–9.
Wang SS, Wu K, Qian Q, Liu Q, Li Q, Pan YJ, Ye YF, Liu XY, Wang J, Zhang JQ, Li S, Wu YJ, Fu XD. Non-canonical regulation of SPL transcription factors by a human OTUB1-like deubiquitinase defifines a new plant type rice associated with higher grain yield. Cell Res. 2017;27(9):1142–56.
Fan CC, **ng YZ, Mao HL, Lu TT, Han B, Xu CG, Li XH, Zhang QF. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet. 2006;112(6):1164–71.
Ishimaru K, Hirotsu N, Madoka Y, Murakami N, Hara N, Onodera H, Kashiwagi T, Ujiie K, Shimizu BI, Onishi A, Miyagawa H, Katoh E. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat Genet. 2013;45(6):707–11.
Wang SK, Li S, Liu Q, Wu K, Zhang JQ, Wang SS, Wang Y, Chen XB, Zhang Y, Gao CX, Wang F, Huang HX, Fu XD. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet. 2015;47(8):949–54.
Wang YX, **ong GS, Hu J, Jiang L, Yu H, Xu J, Fang YX, Zeng LJ, Xu E, Xu J, Ye WJ, Meng XB, Liu RF, Chen HQ, **g YH, Wang YH, Zhu XD, Li JY, Qian Q. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat Genet. 2015;47(8):944–8.
Si LZ, Chen JY, Huang XH, Gong H, Luo JH, Hou QQ, Zhou TY, Lu TT, Zhu JJ, Shangguan YY, Chen EW, Gong CX, Zhao Q, **g YF, Zhao Y, Yan L, Cui LL, Fan DL, Lu YQ, Weng QJ, Wang YC, Zhan QL, Liu KY, Wei XH, An KS, An GH, Han B. OsSPL13 controls grain size in cultivated rice. Nat Genet. 2016;48(4):447–56.
Hu J, Wang YX, Fang YX, Zeng LJ, Xu J, Yu HP, Shi ZY, Pan JJ, Zhang D, Kang SJ, Zhu L, Dong GJ, Guo LB, Zeng DL, Zhang GH, **e LH, **ong GS, Li JY, Qiang Q. A rare allele of GS2 enhances grain size and grain yield in rice. Mol Plant. 2015;8(10):1455–65.
Che RH, Tong HN, Shi BH, Liu YQ, Fang SR, Liu DP, **ao YH, Hu B, Liu LC, Wang HR, Zhao MF, Chu CC. Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat Plants. 2015;2:15195.
Duan PG, Ni S, Wang JM, Zhang BL, Xu R, Wang YX, Chen HQ, Zhu XD, Li YH. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat Plants. 2015;2:15203.
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–30.
Liu JF, Chen J, Zheng XM, Wu FQ, Lin QB, Heng YQ, Tian P, Cheng ZJ, Yu XW, Zhou KN, Zhang X, Guo XP, Wang JL, Wang HY, Wan JM. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat Plants. 2017;3:17043.
Zhu ZF, Tan LB, Fu YC, Liu FX, Cai HW, **e DX, Wu F, Wu JZ, Matsumoto T, Sun CQ. Genetic control of inflorescence architecture during rice domestication. Nat Commun. 2013;4:2200.
Kinoshita T, Takamure I. Linkage studies by the use of backcross data in rice. Genetical studies on rice plant, LXXXVIII. J Fac Agr Hokkaido Univ. 1986;61:445–55.
Mitra S, Ganguli P. Some observations on the characters of wild rice hybrids. Indian J Agric Sci. 1932;2:271–9.
Luo JJ, Hao W, ** J, Gao JP, Lin XH. Fine map** of Spr3, a locus for spreading panicle from african cultivated rice (Oryza glaberrima Steud). Mol Plant. 2008;1(5):830–8.
Sanchez A, Khush G. A new gene for spreading panicle in rice. Rice Genet Newsl. 1997;14:47–8.
Xu Y, Zhou J, Hu F, Xu P, Deng X, Li J, Ren G, Tao D. Spr5(t) a spreading panicle gene from Oryza glaberrima. Rice Genet Newsl. 2008;25:16–7.
Chen PP, Ye SH, Lu YT. Characteristics and mutation gene map** of late japonica rice Zhe**g 22 loose spike mutant spr8. Acta Agric Nucl Sin. 2013; 27: 1–8.
Nakamura K, Izumiyama N, Ohtsubo K, Koiso Y, Iwasaki S, Sonoda R, Fujita Y, Yaegashi H, Sato Z. Lupinosis”-like lesions in mice caused by ustiloxin, produced by Ustilaginoidea virens: a morphological study. Nat Toxins. 1994;2(1):22–8.
Song TQ, Zhang Y, Zhang Q, Zhang X, Shen DY, Yu JJ, Yu MN, Pan XY, Cao HJ, Yong ML, Qi ZQ, Du Y, Zhang RS, Yin XL, Qiao JQ, Liu YZ, Liu WD, Sun WX, Zhang ZG, Wang YC, Dou DL, Ma ZC, Liu YF. The N- terminus of an Ustilaginoidea virens ser- thr- rich glycosylphosphatidylinositol- anchored protein elicits plant immunity as a MAMP. Nat Commun. 2021;12(1):2451.
Chen XY, Duan YH, Qiao FG, Liu H, Huang JB, Luo CX, Chen XL, Li GT, **e KB, Hsiang T, Zheng L. A secreted fungal effector suppresses rice immunity through host histone hypoacetylation. New Phytol. 2022;235(5):1977–94.
Long WX, Yuan ZQ, Fan FF, Dan D, Pan GJ, Sun HM, Zhang ZH, Li NW, Li SQ. Genome-wide association analysis of resistance to rice false smut. Mol Breed. 2020;40:46.
He NQ, Yang DW, Zheng XH, Huang FH, Cheng CP, Ye N. Improving blast resistance of R20 by molecular marker-assisted selection of the Pigm-1 gene. Acta Agric Nucl Sin. 2022;36(2):245–50.
Yang DW, Li SP, **ao YP, Lu L, Zheng ZC, Tang DZ, Cui HT. Transcriptome analysis of rice response to blast fungus identified core genes involved in immunity. Plant Cell Environ. 2021;44(9):3103–21.
Song JH, Wei W, Lv B, Lin Y, Yin WX, Peng YL, Schnabel G, Huang JB, Jiang DH, Luo CX. Rice false smut fungus hijacks the rice nutrients supply by blocking and mimicking the fertilization of rice ovary. Environ Microbiol. 2016;18(11):3840–9.
Tang CS, Gao JZ, Cao GP, Huang SH, Liu RM, Liu CL, **e W, Luo XY, **ao Q. Reasearch and application of classification standard of rice false smut. Plant Prot. 2001;27:18–21.
Yang DW, Ye XF, Zheng XH, Cheng CP, Ye N, Huang FH. Development and evaluation of chromosome segment substitution lines carrying overlap** chromosome segments of the whole wild rice genome. Front Plant Sci. 2016;7:01737.
Panaud O, Chen X, Mccouch S. Development of microsatellite and characterization of simple sequence length polymorphism (SSLP) in rice (Oryza sativa L). Mol Gen Genet. 1996;252(5):597–607.
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L. Mapmaker: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987;1:174–81.
Rahman M, Chu S, Choi MS, Qian YL, Jiang WZ, Piao RH, Khanam S, Cho YI, Jeung JU, Jena K, Koh HJ. Identification of QTLs for some agronomic traits in rice using an introgression line from Oryaza minuta. Mol Cell. 2007;24(1):16–26.
**e KB, Zhang JW, Yang YN. Genome-wide prediction of highly specific guide RNA spacers for CRISPR-Cas9-mediated genome editing in model plants and major crops. Mol Plant. 2014;7(5):923–6.
Ma XL, Chen LT, Zhu QL, Chen YL, Liu YG. Rapid decoding of sequence-specific nuclease-induced heterozygous and biallelic mutations by direct sequencing of PCR products. Mol Plant. 2015;8(8):1285–7.
Xu GY, Greene GH, Yoo HJ, Liu LJ, Marques J, Motley J, Dong XN. Global translational reprogramming is a fundamental layer of immune regulation in plants. Nature. 2017;545(7655):487–90.
Zhang Y, Zhao JH, Li YL, Yuan ZJ, He HY, Yang HH, Qu HY, Ma CY, Qu SH. Transcriptome analysis highlights defense and signaling pathways mediated by Rice pi21 gene with partial resistance to Magnaporthe oryzae. Front Plant Sci. 2016;7:1834.
Zhang YH, ** YY, Gong Q, Li Z, Zhao LH, Han X, Zhou JH, Li FG, Yang ZE. Mechanismal analysis of resistance to verticillium dahliae in upland cotton conferred by overexpression of rpl18a-6 (ribosomal protein l18a-6). Ind Crops Prod. 2019;141:111742.
Carlson CH, Fiedler JD, Naraghi SM, Nazareno ES, Ardayfio NK, McMullen MS, Kianian SF. Archetypes of inflorescence: genome-wide association networks of panicle morphometric, growth, and disease variables in a multiparent oat population. Genetics. 2022;15(128):36106985.
Eizenga GC, Jia MH, Jackson AK, Boykin DL, Ali ML, Shakiba E, Tran NT, McCouch SR, Edwards JD. Validation of yield component traits identified by genome-wide association map** in a tropical japonica × tropical japonica rice biparental map** population. Plant Genome. 2019;12(1):30951093.
Lu MH, Liu WC, Zhu F. 2018. Epidemic law and control technique of rice false smut in recent years. China Plant Prot. 2018; 38: 44–47
Sun WX, Fan J, Fang AF, Li YJ, Tariqjaveed M, Li DY, Hu DW, Wang WM. Ustilaginoidea virens: insights into an Emerging Rice Pathogen. Annu Rev Phytopathol. 2020;58:363–85.
Vaughan D, Morishima H, Kadowaki K. Diversity in the Oryza genus. Curr Opin Plant Biol. 2003;6(2):139–46.
Grillo M, Li C, Fowlkes AM, Briggeman TM, Zhou AL, Schemske DW, Sang T. Genetic architecture for the adaptive origin of annual wild rice, Oryza mvara. Evolution. 2009;63(4):870–83.
Acknowledgements
Not applicable.
Funding
The work was supported by the Special Fund for Agro-scientific Research in the Public Interest of Fujian Province (2023R11010021-1), the Fujian Provincial Natural Science Foundation (No. 2023J01141340, 2022J01450 and 2021J01471), the Free Exploration Project of Fujian Academy of Agricultural Sciences (ZYTS202308 and ZYTS2023006), Major Science and Technology Projects of Fujian Province (No. 2020NZ08016), Science and Technology Innovation Team (No. CXTD2021005-3), Youth Science and Technology Innovation Team (No. CXTD2021001), 5511 Collaborative Engineering Project (No. KXXYJBG0021), and the 100 Talent Plans of Fujian Province.
Author information
Authors and Affiliations
Contributions
All the authors contributed to the conception and design of the study. DY planned and performed the experiments and data collection and wrote the manuscript with input from all authors. The collection and analysis of the data were performed by NH, FH, LL and XW. The first draft of the manuscript was written by DY, NH. NH prepared Fig. 1, FH prepared Fig. 2, LL prepared Fig. 3 and XW prepared Fig. 4. QQL revised the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, N., Huang, F., Lu, L. et al. SPR9 encodes a 60 S ribosomal protein that modulates panicle spreading and affects resistance to false smut in rice (Oryza sativa. L). BMC Plant Biol 23, 205 (2023). https://doi.org/10.1186/s12870-023-04172-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04172-4