
Abstract
Background
Guizhou black goat is one of the indigenous black goat breeds in the southwest region of Guizhou, China, which is an ordinary goat for mutton production. They are characterized by moderate body size, black coat, favorite meat quality with tender meat and lower odor, and tolerance for cold and crude feed. However, little is known about the genetic characteristics or variations underlying their important economic traits.
Results
Here, we resequenced the whole genome of Guizhou black goat from 30 unrelated individuals breeding in the five core farms. A total of 9,835,610 SNPs were detected, and 2,178,818 SNPs were identified specifically in this breed. The population structure analysis revealed that Guizhou black goat shared a common ancestry with Shaanbei white cashmere goat (0.146), Yunshang black goat (0.103), Iran indigenous goat (0.054), and Moroccan goat (0.002). However, Guizhou black goat showed relatively higher genetic diversity and a lower level of linkage disequilibrium than the other seven goat breeds by the analysis of the nucleotide diversity, linkage disequilibrium decay, and runs of homozygosity. Based on FST and θπ values, we identified 645, 813, and 804 selected regions between Guizhou black goat and Yunshang black goat, Iran indigenous goat, and cashmere goats. Combined with the results of XP-EHH, there were 286, 322, and 359 candidate genes, respectively. Functional annotation analysis revealed that these genes are potentially responsible for the immune response (e.g., CD28, CD274, IL1A, TLR2, and SLC25A31), humility-cold resistance (e.g., HBEGF, SOSTDC1, ARNT, COL4A1/2, and EP300), meat quality traits (e.g., CHUK, GAB2, PLAAT3, and EP300), growth (e.g., GAB2, DPYD, and CSF1), fertility (e.g., METTL15 and MEI1), and visual function (e.g., PANK2 and NMNAT2) in Guizhou black goat.
Conclusion
Our results indicated that Guizhou black goat had a high level of genomic diversity and a low level of linkage disequilibrium in the whole genome. Selection signatures were detected in the genomic regions that were mainly related to growth and development, meat quality, reproduction, disease resistance, and humidity-cold resistance in Guizhou black goat. These results would provide a basis for further resource protection and breeding improvement of this very local breed.
Similar content being viewed by others
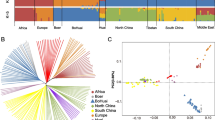


Background
The goat (Capra hircus) is a domesticated species of goat-antelope typically kept as livestock. It is one of the oldest domesticated species of animal, according to archaeological evidence that its earliest domestication occurred 10,000 calibrated calendar years ago in Iran [1]. With the domestication of goats, both natural and artificial selection led to the formation of breeds with distinct phenotypic characteristics including morphological, physiological, and adaptive traits. There are 557 domesticated goat breeds distributed all over the world [2, 3], and 58 indigenous breeds adapted to different agroclimatic conditions in China [4]. Selection signatures, as selective imprints such as genetic polymorphism reduction and linkage disequilibrium left on the genome by natural and/or artificial selection, can affect the agronomic and adaptive traits of domesticated animals and have been well detected by whole genome sequencing (WGS) [5,6, High-throughput sequencing generated genomic data for 30 GZB (Supplementary Table S1) at an average sequencing depth of approximately 15.3-fold. It was jointly genotyped with 79 (Supplementary Fig. 1, Supplementary Table S2) publicly available genomic data from eight representative populations, including Iran indigenous goat (IIG, n = 15), South Korean goat (SKG, n = 12), Moroccan goat (MG, n = 12), Yunshang black goat (YBG, n = 11), French goat (FG, n = 10), Shaanbei white cashmere goat (SCG, n = 9), Wild goat (WG, n = 6), and Tibetan goat (TG, n = 4). The total number of SNPs detected within the populations was shown in Supplementary Table S3. Then we annotated 9,835,610 biallelic SNPs that were discovered in 30 GZB, and GZB had the highest number of SNPs among nine populations (Supplementary Table S3). Functional annotation of the polymorphic sites revealed that the vast majority of SNPs were present in either intergenic regions (65.63%) or intronic regions (27.81%). Exons contained 0.66% of the total SNPs with 46,767 (42.61%) nonsynonymous SNPs and 61,917 (56.41%) synonymous SNPs (Fig. 1, Supplementary Table S4). We also found that the shared SNP counts of nine goat populations were 2,783,053 (Supplementary Fig. 2a), while the unique SNP counts of GZB weres 2,178,818 (Supplementary Fig. 2b). Statistics regarding the whole genome SNP variant types of GZB using ANNOVAR. Plot of the total variant annotation and coding consequence variant annotation To explore relatedness among GZB and other goat populations distributed worldwide, we conducted ADMIXTURE, neighbor-joining (NJ) tree, principal component analysis (PCA), and Maximum likelihood (ML) tree using whole-genome SNP data (Fig. 2). The ADMIXTURE analysis revealed K = 3 (cross-validation error = 0.52634) as the most likely number of genetically distinct populations for nine goat populations (Supplementary Table S5). When K = 8, some GZB showed clear evidence of genetic heterogeneity with shared genome ancestry with SCG (0.146), YBG (0.103), IIG (0.054), and MG (0.002) genetic background (Fig. 2a, Supplementary Table S6). The NJ tree recapitulated these findings and showed that the genetic distance between GZB and other goat populations became farther with the geographical distance. GZB was found to be genetically closer to YBG (Fig. 2b). The PCA (Fig. 2c) showed similar results with NJ tree, which together revealed genetic differences between GZB and other goat populations at the overall genomic level. ML tree analysis showed that when the number of migration events was seven, GZB had gene flow from SCG, and flowed out to TG (Fig. 2d). Population Structure of Guizhou Black Goat and its relationship with other eight goat populations in the world. a Model-based clustering of goat populations using ADMIXTURE. The length of each colored segment represents the proportion of the individual’s genome from K = 3 and K = 8 ancestral populations. The population names are at the top of the figure. b Neighbor-joining phylogenetic tree of the nine goat populations. The scale bar represents proportional to similarity (p distance). c Principal component analysis of nine goat populations. Different colored lines or points represent different categories. GZB (Guizhou black goat), YBG (Yunshang black goat), SCG (Shaanbei white cashmere goat), TG (Tibetan goat), SKG (South Korean goat), IIG (Iran indigenous goat), WG (Wild goat, Capra aegagrus), MG (Moroccan goat), and FG (French goat). d ML tree of nine goat populations with migration edges = 7 To examine the degree of nucleotide sequence variation among individuals in each goat population, nucleotide diversity was calculated. The results showed that nucleotide diversity was the highest in YBG (0.001376), tightly followed by GZB (0.001352) (Fig. 3a). In contrast, we observed a lower value of LD in GZB, following IIG closely (Fig. 3b). a Genome-wide distribution of nucleotide diversity of each population in 100 kb windows with 10 kb steps. The horizontal line inside the box indicates the median of this distribution; box limits indicate the first and the third quartiles, and points show outliers. Data points outside the whiskers can be considered outliers. b Genome-wide average LD decay estimated from each population Based on the results of population genetic structure, distinct agronomic traits, and environmental characteristics of production areas, the goat populations were regrouped into three representative populations: Yunshang black goat (muttony goat breed), Iran indigenous goat (living in arid or semi-arid areas), and Cashmere goat (CG, Shaanbei white cashmere goat and Tibetan goat for cashmere production). Combined FST, θπ, and XP-EHH, we detect genomic regions associated with selection in the GZB with YBG, IIG, and CG, respectively. And we selected the top 1% of signals as candidate regions. The 645 selected regions (blue points) were detected and 252 candidate genes were extracted in the GZB vs. YBG comparison (Fig. 4a-c, Supplementary Fig. 3, and Supplementary Table S7). By adding the XP-EHH to detect among-population selection signals, we obtained 34 selected genes different from the results of the above two methods (Supplementary Fig. 4a). And 258 GO terms and 22 KEGG pathways were significantly enriched (P < 0.05, Fig. 5a-b, Supplementary Table S8-S9). Of which, growth/development-related terms have a high rate of occurrence. The Wnt signaling pathway (P = 0.001) is indispensable in the growth and development, involving PRICKLE2, PPP3R1, CXXC4, RBX1, EP300, and ROR1. Ten GO terms (e.g., osteoblast differentiation, growth factor activity, and limb development) are also significantly enriched in growth/development (P < 0.05). In addition, a region of 0.29 Mb on chromosome 3 containing ENSCHIG00000006864 (novel gene, RNA gene, lncRNA) was strongly selected by FST (average FST = 0.604) and θπ ratio (average θπ ratio = 17.1) (Fig. 4d). Moreover, we noticed METTL15, which is related to mitochondrial rRNA methylation (rRNA base methylation and mitochondrial matrix), showing a strong positive selection signal in GZB (Fig. 4e). A missense mutation (rs648661574, c.A60C, p.E20D) was found at METTL15 gene. This mutation presented a huge divergence between GZB (allele C frequency = 0.9) and YBG (allele A frequency = 1). Analysis of the signatures of positive selection in the genome of GZB compared to YBG. a Distribution of θπ ratios (θπ, YBG/θπ, GZB) and FST values, which are calculated in 100 kb windows sliding in 10 kb steps. Data points located to the right of the vertical dashed lines, respectively (corresponding to the 1% right tail of the empirical θπ ratio distribution, where the θπ ratio is 2.274), and above the horizontal dashed line (the 1% right tail of the empirical FST distribution, where FST is 0.3506) were identified as selected regions for GZB (blue points). b Manhattan plot of selective sweeps using θπ ratio in GZB vs. YBG. Solid blue line represented the threshold of the top 1% θπ ratios. c Manhattan plot of selective sweeps using FST in GZB vs. YBG. Solid blue line represented the threshold of the top 1% FST values. d-e Example of genes with strong selective sweep signals in GZB. θπ ratio and FST values are plotted using a 10 kb sliding window. Gray rectangle regions were termed as regions with strong selective sweep signals for GZB. The boundaries of ENSCHIG00000006864 and METTL15 genes are marked in red GO and KEGG pathway enrichment analysis shows significant (P < 0.05) terms, pathways, and associated genes in GZB vs. YBG comparison. a The Sankey-Dot plot of the 16 significant KEGG pathways. b The Sankey-Dot plot of the Top 20 GO terms. The size of circles for each pathway represents counts of associated genes. The color of the circles indicates the P-value In the selection signals and selective sweep analysis between GZB and IIG, 813 selected regions and 324 candidate genes were scanned by FST and θπ (Supplementary Fig. 3, Supplementary Fig. 5a-c, and Supplementary Table S7). We obtained 32 selected genes by adding the XP-EHH (Supplementary Fig. 4b). In KEGG pathway enrichment analysis, the top 1 was osteoclast differentiation (P = 0.001), and 17 (17/32) immune-related pathways (e.g., rheumatoid arthritis, Human papillomavirus infection, and type I diabetes mellitus) were enriched (P < 0.05, Supplementary Table S10). The selected gene IL1A (Supplementary Fig. 5d) is involved in six of these pathways and is associated with disease. In GO enrichment analysis, we got 281 significant terms (P < 0.05, Supplementary Table S11). There were several significant terms related to environmental adaptation, such as cellular response to UV and cellular response to heat. In addition, a region of 0.24 Mb on chromosome 4 (containing DNAJC2, PMPCB, PSMC2, RELN, and SLC26A5) was strongly selected by FST and θπ ratio (Supplementary Fig. 5e), and a nonsynonymous SNV (novel variant, c.A4G, p.T2A) was found in the PMPCB gene. Allele G displayed an abundant distribution (frequency = 1) in GZB, whereas it showed an opposite pattern (frequency = 0) in IIG. A total of 804 selected regions and 359 candidate genes were found in the comparison of GZB and CG (Supplementary Fig. 3, Supplementary Fig. 4c, Supplementary Fig. 6a-c, and Supplementary Table S7), and eleven of them were significantly enriched six fiber-related terms (e.g., keratin filament, cornification, elastic fiber, stress fiber, and regulation of keratinocyte proliferation) (P < 0.05, Supplementary Table S12). Moreover, two missense mutations (rs667703315, c.G1511A, p.S504N. rs649013003, c.A2680G, p.N894D) were found at the KRT79 and PRKD1 genes among eleven genes, respectively (Supplementary Fig. 6d-e). As one of the peculiar selected genes in this comparison group, JAK2, containing a nonsynonymous SNV (rs647159917, c.G1573A, p.A525T) is involved in multiple immune-related pathways (e.g., influenza A, Th1 and Th2 cell differentiation, and Th17 cell differentiation) (P < 0.05, Supplementary Table S13). We inspected 11 selected SNPs (Supplementary Table S14) in candidate functional genes below from 30 individuals obtained by the Sanger sequencing approach, giving an overall validation rate of 99.39%. Overall, the results indicated confidence in the correct rate of variant calling of SNP. Characterizing population structure and genetic diversity is essential for the revelation of evolutionary history, understanding of environmental adaptation, conservation and utilization of germplasm resources, and investigation of phylogenetic relationships. In this study, we performed a whole-genome resequencing analysis of 30 GZB. This is the hitherto most comprehensive data set on the population genetic structure of GZB. Neither the number of individuals nor the depth of sequencing used in previous studies is comparable to this study. We found that GZB had the highest number of SNPs among the nine populations. This may be related to the number of samples and the depth of whole genome resequencing. Then we explored the population genetic structure of GZB in the context of the goat populations with potential ancestors and identified useful nonsynonymous SNPs that involved local adaptation and agronomic traits. As shown in Fig. 2a, nearly half (13) of the GZB contained ancestral contributions from SCG (~ 14.6%), YBG (~ 10.3%), and IIG (~ 5%), and more than half (17) of the GZB with pure genetic background seemed to have originated locally in Guizhou, China. In other words, we needed to integrate more in-depth information to confirm the origin of GZB. Meanwhile, the habitation and relationship of GZB was nearby to the YBG in southwest China (Fig. 2b-c). GZB and YBG (mean θπ = 0.001376) showed a similar level of nucleotide diversity, which may be related to their similar genetic background (Fig. 3a). The relatively high level of genomic diversity found in GZB might reflect the stronger selection pressure and longer selection history. In addition, the patterns of LD decay in each population were largely consistent with the results of nucleotide diversity. The above results confirmed that the GZB harbored fewer variants, lower linkage decay, and higher nucleotide diversity comparable to the other native populations, suggesting unique genetic characteristics. The typical characteristic of GZB is moderate in body size, approximately 55 cm in height, and ~ 30 kg in weight at one year old [14]. Body size is a key factor in determining mutton production. When analyzing the selection signatures of GZB with the bigger YBG (weighing 46 kg at one year old), several positively selected genes were detected associate with growth (SUOX, CSF1, CHUK, DPYD, and GDF2) and fatty acid metabolism (GAB2, SMOX, and GOT2). Sulfite oxidase (SUOX) plays an important role in bovine bone development [15]. CSF1 is involved in the fast growth rate of large white pigs at an early stage [16] and is an essential growth factor for osteoclast progenitors and an important regulator for bone resorption [17]. A previous study suggests that CHUK has an intrinsic cell-autonomous role in chondrocytes that controls chondrocyte phenotype and affects ontogeny [18]. DPYD is related not only to muscle growth but also to fat deposition [19]. Previous studies have demonstrated that GDF2 is the most potent bone morphogenetic protein that can be used to induce bone formation from mesenchymal stem cells both in vitro and in vivo, through a comprehensive analysis of osteogenic activity [20,21,22]. Gab2 plays an important role in regulating adipocyte maturation, differentiation, and function by using mouse primary or immortalized brown preadipocytes in vitro [23]. SHOX is considered to be involved in the physiological processes of sheep growth and carcass composition traits [24]. GOT2 can affect pork quality by participating in aromatic amino acid metabolism [56] by default parameters, such as samtools view -bS A.sam > A.sort.bam. Duplicates were removed by the MarkDuplicates module in GATK v.4.3.0.0 [57] with command ‘gatk –java-options "-Xmx16g -Djava.io.tmpdir = ./tmp" MarkDuplicates -I A.sort,bam -M A.metrics –CREATE_INDEX -O A.sort.MarkDup.bam’. SNPs and Indels were called from the bam files by the GATK HaplotypeCaller module with the GATK best-practice recommendations [57]. The recommended command was like gatk –java-options "-Xmx4g" HaplotypeCaller -R ARS1.fa -I A.sort.MarkDup.bam -O A.g.vcf.gz. Raw GVCFs with the samples called individually were merged using the CombineGVCFs and genotyped by the GenotypeGVCFs. We then extracted and filtered SNPs using the GATK module SelectVariants. The recommended command was like gatk SelectVariants -R ARS1.dna.toplevel.fa -V output.vcf.gz –select-type-to-include SNP -O raw_snps_genotype.vcf. To avoid potential false-positive calls, we implemented "VariantFiltering" of the GATK for the selected SNPs using the best practice parameters "QUAL > 30.0 || QD < 2.0 || FS > 60.0 || MQ < 40.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0". We then filtered out nonbiallelic SNPs. After the quality screening, all the identified SNPs were further annotated using ANNOVAR [58] based on the gene annotations of the goat reference genome ARS1. Locations for SNPs in various genic and intergenic regions and synonymous or nonsynonymous SNPs in exonic regions were annotated. We pruned the SNPs in high levels of pair-wise LD using PLINK [59] with the parameter (–indep-pairwise 50 10 0.2) to perform principal component analysis (PCA) and ADMIXTURE analysis. As PCA, the first two eigenvectors were plotted in the ggplot2 package under the R platform. Population structure analysis was carried out using ADMIXTURE v1.3 [60] with kinship (K) set from 2 to 9. Gene flow analysis was performed using Treemix with m = 5 and i = 10. The unrooted NJ tree was constructed by PLINK using the matrix of pairwise genetic distances and visualized with MEGA X [61] and FigTree v1.4.4 (http://tree.bio.ed.ac.uk). Construction and visualization of the ML tree was done by Treemix [62] v1.1.3. The squared correlation (r2) between any two loci was calculated to evaluate Linkage disequilibrium (LD) decay using the PopLDdecay v3.41 [63]. A sliding-window approach (100 kb windows sliding in 10 kb step size) was applied to quantify polymorphism levels (θπ, the ratio of nucleotide diversity) and pairwise genetic differentiation (FST) between GZB and other goat populations. The programs were used to calculate θπ and FST: vcftools –vcf /data/SNP.vcf –keep /data/GZB.txt –window-pi 100,000 –window-pi-step 10,000 –out /data/GZB_IIG, vcftools –vcf /data/SNP.vcf –keep /data/IIG.txt –window-pi 100,000 –window-pi-step 10,000 –out /data/cll/IIG_GZB; vcftools –vcf /data/SNP.vcf –weir-fst-pop /data/GZB.txt –weir-fst-pop /data/IIG.txt –out /data/GZB_IIG –fst-window-size 100,000 –fst-window-step 10,000. XP-EHH was calculated by chromosome with command: selscan [64] –xpehh –vcf GZB.chr k.vcf –vcf-ref YBG.chr k.vcf –map chr k.MT.map.distance –out chr k.GZB_YBG.out. To detect regions with significant signatures of selective sweeps, we divided the 4 goat populations (except for Wild goat, Morocco goat, French goat, and South Korean goat) into three reference populations, namely CG (Cashmere goat, 9 Shaanbei white cashmere goat, 4 Tibetan goat), IIG (15 Iran indigenous goat), YBG (11 Yunshang black goat). To uncover selection signatures of GZB, we calculated pairwise FST and θπ in 100 kb sliding windows with a step size of 10 kb across the autosomes between GZB and YBG, IIG, or CG populations, respectively. The windows with high values of θπ ratio and FST, representing the top 1% of all windows, were determined as the selected regions. Kyoto Encyclopedia of Genes and Genomes (KEGG) [65,66,67] pathways and Gene Ontology (GO) terms were analyzed based on the candidate genes via FST and θπ methods using KOBAS-intelligence [68] to investigate the biological enrichment of genes under selective pressure. The GO terms and KEGG pathways were considered to be significantly enriched only when the P-value was less than 0.05. To check the confidence of SNPs called, we randomly validated 11 SNPs in specific genes from 30 individuals that were genotyped by PCR and Sanger sequencing. The primers used for PCR were designed with DNAMAN v9.0.1.116 (Lynnon Biosoft, USA). The PCR reactions were carried out in 50 μL volume containing 25 μL of 2 × taq PCR Master Mix (TIANGEN Biotech, Bei**g, China), 2 μL (10 pmol/mL) for each forward and reverse primer (Supplementary table S14), 2.5 μL DNA templates (30-100 ng/mL), and the remainder supplied with dd H2O. The reactions were performed by a BIO-RAD T100 Thermal Cycler with conditions of an initial denaturation at 95 °C for 5 min, followed by 35 cycles at 95 °C for 30 s, annealing at 58/61/65 °C for 30 s and extension at 72 °C for 45 s, and then a final extension at 72 °C for 5 min. All the reads were assessed manually and genotypes of each site were identified by the Sanger sequencing peaks. Subsequently, we compared genotypes of each site identified by whole-genome resequencing and obtained by the Sanger sequencing for the same individuals.Results
Sequencing and identification of SNPs

Population structure of Guizhou black goat and other eight goat populations

Genetic diversity, and linkage disequilibrium of nine goat populations

Detection of selection signals and selective sweep in GZB


Variant accuracy
Discussion
Phylogenetic and population genetic analyses
Calculation of θπ, F
ST, and XP-EHH
Identification of selected regions
Gene functional enrichment analysis
SNP validation
Availability of data and materials
Sequences were private from ENA with the Bioproject accession numbers PRJEB67694.
Abbreviations
- GZB:
-
Guizhou black goat
- YBG:
-
Yunshang black goat
- CG:
-
Cashmere goat
- IIG:
-
Iran indigenous goat
- TG:
-
Tibetan goat
- SCG:
-
Shaanbei white cashmere goat
- SKG:
-
South Korean goat
- WG:
-
Wild goat
- FG:
-
French goat
- MG:
-
Morocco goat
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole-genome sequencing
- θπ :
-
Nucleotide diversity
- F ST :
-
Fixation index
- XP-EHH:
-
The Cross-Population Extended Haplotype Homozygousity
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- NJ:
-
Neighbor-joining
- PCA:
-
Principle component analysis
- LD:
-
Linkage disequilibrium
References
Zeder MA, Hesse B. The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science (New York, NY). 2000;287(5461):2254–7.
Wang X, Liu J, Zhou G, Guo J, Yan H, Niu Y, Li Y, Yuan C, Geng R, Lan X, et al. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci Rep. 2016;6:38932.
Guo J, Tao H, Li P, Li L, Zhong T, Wang L, Ma J, Chen X, Song T, Zhang H. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci Rep. 2018;8(1):10405.
Skapetas B, Bampidis VJLRfRD: Goat production in the world: Present situation and trends. 2016.
Yang BG, Yuan Y, Zhou DK, Ma YH, Mahrous KF, Wang SZ, He YM, Duan XH, Zhang WY. E G: Genome-wide selection signal analysis of Australian Boer goat reveals artificial selection imprinting on candidate genes related to muscle development. Anim Genet. 2021;52(4):550–5.
Li R, Yang P, Dai X, Asadollahpour Nanaei H, Fang W, Yang Z, Cai Y, Zheng Z, Wang X, Jiang Y. A near complete genome for goat genetic and genomic research. Genet Sel Evol. 2021;53(1):74.
**ong J, Bao J, Hu W, Shang M, Zhang L. Whole-genome resequencing reveals genetic diversity and selection characteristics of dairy goat. Front Genet. 2022;13:1044017.
Cai Y, Fu W, Cai D, Heller R, Zheng Z, Wen J, Li H, Wang X, Alshawi A, Sun Z, et al. Ancient Genomes Reveal the Evolutionary History and Origin of Cashmere-Producing Goats in China. Mol Biol Evol. 2020;37(7):2099–109.
** M, Lu J, Fei X, Lu Z, Quan K, Liu Y, Chu M, Di R, Wei C, Wang H: Selection Signatures Analysis Reveals Genes Associated with High-Altitude Adaptation in Tibetan Goats from Nagqu, Tibet. Animals (Basel) 2020, 10(9).
Wang JJ, Li ZD, Zheng LQ, Zhang T, Shen W, Lei CZ. Genome-wide detection of selective signals for fecundity traits in goats (Capra hircus). Gene. 2022;818:146221.
Tao L, He XY, Jiang YT, Lan R, Li M, Li ZM, Yang WF, Hong QH, Chu MX. Combined approaches to reveal genes associated with litter size in Yunshang black goats. Anim Genet. 2020;51(6):924–34.
Li Y, Gong Y, Zhang Z, Li L, Liu X, He X, Zhao Q, Pu Y, Ma Y, Jiang L: Whole-genome sequencing reveals selection signals among Chinese, Pakistani, and Nepalese goats. Journal of genetics and genomics = Yi chuan xue bao 2023, 50(5):362–365.
Chen Q, Huang Y, Wang Z, Teng S, Hanif Q, Lei C, Sun J. Whole-genome resequencing reveals diversity and selective signals in Longlin goat. Gene. 2021;771:145371.
Tan XS, Wang Y, ** M, Wang Q, Gong F, Liu C, Chen CH, Gan L, Mao FX. Establishment of regression model of body weight and body size of Guizhou black goats (in Chinese). Guizhou J Anim Husbandry Vet Med. 2023;47(1):3.
Drögemüller C, Tetens J, Sigurdsson S, Gentile A, Testoni S, Lindblad-Toh K, Leeb T: Identification of the bovine Arachnomelia mutation by massively parallel sequencing implicates sulfite oxidase (SUOX) in bone development. PLoS genetics 2010, 6(8).
Cai C, Li M, Zhang Y, Meng S, Yang Y, Gao P, Guo X, Cao G, Li B. Comparative Transcriptome Analyses of Longissimus thoracis Between Pig Breeds Differing in Muscle Characteristics. Front Genet. 2020;11:526309.
Zhong L, Lu J, Fang J, Yao L, Yu W, Gui T, Duffy M, Holdreith N, Bautista CA, Huang X, et al: Csf1 from marrow adipogenic precursors is required for osteoclast formation and hematopoiesis in bone. Elife 2023, 12.
Culley KL, Lessard SG, Green JD, Quinn J, Chang J, Khilnani T, Wondimu EB, Dragomir CL, Marcu KB, Goldring MB, et al. Inducible knockout of CHUK/IKKα in adult chondrocytes reduces progression of cartilage degradation in a surgical model of osteoarthritis. Sci Rep. 2019;9(1):8905.
Chen G, Cheng X, Shi G, Zou C, Chen L, Li J, Li M, Fang C, Li C. Transcriptome Analysis Reveals the Effect of Long Intergenic Noncoding RNAs on Pig Muscle Growth and Fat Deposition. Biomed Res Int. 2019;2019:2951427.
Luther G, Wagner ER, Zhu G, Kang Q, Luo Q, Lamplot J, Bi Y, Luo X, Luo J, Teven C, et al. BMP-9 induced osteogenic differentiation of mesenchymal stem cells: molecular mechanism and therapeutic potential. Curr Gene Ther. 2011;11(3):229–40.
Lamplot JD, Qin J, Nan G, Wang J, Liu X, Yin L, Tomal J, Li R, Shui W, Zhang H, et al. BMP9 signaling in stem cell differentiation and osteogenesis. Am J Stem Cells. 2013;2(1):1–21.
Cui J, Zhang W, Huang E, Wang J, Liao J, Li R, Yu X, Zhao C, Zeng Z, Shu Y, et al. BMP9-induced osteoblastic differentiation requires functional Notch signaling in mesenchymal stem cells. Lab Invest. 2019;99(1):58–71.
Wang X, Zhao Y, Zhou D, Tian Y, Feng G, Lu Z. Gab2 deficiency suppresses high-fat diet-induced obesity by reducing adipose tissue inflammation and increasing brown adipose function in mice. Cell Death Dis. 2021;12(2):212.
Yilmaz O, Kizilaslan M, Arzik Y, Behrem S, Ata N, Karaca O, Elmaci C, Cemal I. Genome-wide association studies of preweaning growth and in vivo carcass composition traits in Esme sheep. J Anim Breed Genet. 2022;139(1):26–39.
Wang H, **a P, Lu Z, Su Y, Zhu W. Time-restricted feeding affects transcriptomic profiling of hypothalamus in pigs through regulating aromatic amino acids metabolism. J Sci Food Agric. 2023;103(3):1578–87.
Schuermans N, El Chehadeh S, Hemelsoet D, Gautheron J, Vantyghem MC, Nouioua S, Tazir M, Vigouroux C, Auclair M, Bogaert E, et al. Loss of phospholipase PLAAT3 causes a mixed lipodystrophic and neurological syndrome due to impaired PPARγ signaling. Nat Genet. 2023;55(11):1929–40.
Rahman SMK, Sasaki S, Uyama T, Hussain Z, Sikder MM, Saiga H, Ohmura-Hoshino M, Ohta KI, Miki Y, Hoshino K, et al. PLAAT1 deficiency alleviates high-fat diet-induced hepatic lipid accumulation in mice. FASEB J. 2023;37(7):e23032.
Wang Z, Chai J, Wang Y, Gu Y, Long K, Li M, ** L: LncPLAAT3-AS Regulates PLAAT3-Mediated Adipocyte Differentiation and Lipogenesis in Pigs through miR-503–5p. Genes 2023, 14(1).
Fei CF, Zhou LQ. Gene mutations impede oocyte maturation, fertilization, and early embryonic development. BioEssays. 2022;44(10):e2200007.
Dong J, Zhang H, Mao X, Zhu J, Li D, Fu J, Hu J, Wu L, Chen B, Sun Y, et al. Novel biallelic mutations in MEI1: expanding the phenotypic spectrum to human embryonic arrest and recurrent implantation failure. Hum Reprod. 2021;36(8):2371–81.
Zhang Y, Li N, Ji Z, Bai H, Ou N, Tian R, Li P, Zhi E, Huang Y, Zhao J, et al. Bi-allelic MEI1 variants cause meiosis arrest and non-obstructive azoospermia. J Hum Genet. 2023;68(6):383–92.
Chappel S. The role of mitochondria from mature oocyte to viable blastocyst. Obstet Gynecol Int. 2013;2013:183024.
Goto S, Konnai S, Hirano Y, Kohara J, Okagawa T, Maekawa N, Sajiki Y, Watari K, Minato E, Kobayashi A, et al. Upregulation of PD-L1 Expression by Prostaglandin E(2) and the Enhancement of IFN-γ by Anti-PD-L1 Antibody Combined With a COX-2 Inhibitor in Mycoplasma bovis Infection. Front Vet Sci. 2020;7:12.
Zheng Q, Hou J, Zhou Y, Yang Y, **e B, Cao X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015;25(10):1121–36.
Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development (Cambridge, England). 2004;131(7):1619–28.
Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354(14):1489–96.
Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Limb and skin abnormalities in mice lacking IKKalpha. Science (New York, NY). 1999;284(5412):313–6.
Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science (New York, NY). 1999;284(5412):316–20.
Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A: The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database : the journal of biological databases and curation 2016, 2016.
Yin J, Guo Y. HOXD13 promotes the malignant progression of colon cancer by upregulating PTPRN2. Cancer Med. 2021;10(16):5524–33.
Coit P, Ognenovski M, Gensterblum E, Maksimowicz-McKinnon K, Wren JD, Sawalha AH. Ethnicity-specific epigenetic variation in naïve CD4+ T cells and the susceptibility to autoimmunity. Epigenetics Chromatin. 2015;8:49.
Lee S. The association of genetically controlled CpG methylation (cg158269415) of protein tyrosine phosphatase, receptor type N2 (PTPRN2) with childhood obesity. Sci Rep. 2019;9(1):4855.
Deng S, Yu K, Jiang W, Li Y, Wang S, Deng Z, Yao Y, Zhang B, Liu G, Liu Y, et al. Over-expression of Toll-like receptor 2 up-regulates heme oxygenase-1 expression and decreases oxidative injury in dairy goats. J Anim Sci Biotechnol. 2017;8:3.
Xu P, Xu X, Fotina H, Fotina T. Anti-inflammatory effects of chlorogenic acid from Taraxacum officinale on LTA-stimulated bovine mammary epithelial cells via the TLR2/NF-κB pathway. PLoS ONE. 2023;18(3):e0282343.
Paul A, Dangi SS, Gupta M, Singh JV, Thakur N, Naskar S, Nanda PK, Mohanty N, Das A, Bandopadhayay S, et al. Expression of TLR genes in Black Bengal goat (Capra hircus) during different seasons. Small Rumin Res. 2015;124:17–23.
Wu ZM, **ang YR, Zhu XB, Shi XD, Chen S, Wan X, Guo J. Icariin represses the inflammatory responses and survival of rheumatoid arthritis fibroblast-like synoviocytes by regulating the TRIB1/TLR2/NF-kB pathway. Int Immunopharmacol. 2022;110:108991.
Liu Y, Cheng H, Wang S, Luo X, Ma X, Sun L, Chen N, Zhang J, Qu K, Wang M, et al. Genomic Diversity and Selection Signatures for Weining Cattle on the Border of Yunnan-Guizhou. Front Genet. 2022;13:848951.
Qiang L, Yang S, Cui YH, He YY. Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing. Autophagy. 2021;17(9):2128–43.
Ågren MS, Litman T, Eriksen JO, Schjerling P, Bzorek M, Gjerdrum LMR: Gene Expression Linked to Reepithelialization of Human Skin Wounds. Int J Mol Sci 2022, 23(24).
Yoon SY, Detmar M. Sostdc1 Secreted from Cutaneous Lymphatic Vessels Acts as a Paracrine Factor for Hair Follicle Growth. Curr Issues Mol Biol. 2022;44(5):2167–74.
Ramayo-Caldas Y, Ballester M, Fortes MR, Esteve-Codina A, Castelló A, Noguera JL, Fernández AI, Pérez-Enciso M, Reverter A, Folch JM. From SNP co-association to RNA co-expression: novel insights into gene networks for intramuscular fatty acid composition in porcine. BMC Genomics. 2014;15:232.
Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, Gjesing AP, Eiberg H, Tuomi T, Almgren P, et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun. 2016;7:11089.
Nguyen T, Zheng M, Knapp M, Sladojevic N, Zhang Q, Ai L, Harrison D, Chen A, Sitikov A, Shen L, et al. Endothelial Aryl Hydrocarbon Receptor Nuclear Translocator Mediates the Angiogenic Response to Peripheral Ischemia in Mice With Type 2 Diabetes Mellitus. Front Cell Dev Biol. 2021;9:691801.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (Oxford, England). 2018;34(17):i884–90.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2009;25(14):1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009;25(16):2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e164.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics. 2011;12:246.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35(6):1547–9.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967.
Zhang C, Dong SS, Xu JY, He WM, Yang TL. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics (Oxford, England). 2019;35(10):1786–8.
Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587-d592.
Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, Wu Y, Zhao L, Liu J, Guo J, et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021;49(W1):W317-w325.
Acknowledgements
We thank Song Derong, Zhou Darong, Chen **a, Huang Yongpeng, and others from the Bijie City Husbandry and Veterinary Science Research Institute of Guizhou Province for their assistance in the sampling process.
Funding
This study was funded by the National Natural Science Foundation of China (No. 30560104), and the Guizhou Provincial Science and Technology Projects (QKHPTRC[2019]5615, QKHRC[2016]4012, QKHNZ-2007–3008, QKHZC[2019]2279). The funding body was not involved in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JW and XR conceived and designed the experiments. XN, SH, SL, and QL contributed to the sample collections. LC performed the analysis of whole-genome resequencing data. LC and YZ performed the sample DNA extraction and the SNPs validation experiment. LC drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Animal Care and Use Committee of Guizhou University following the recommendation of the Regulations for the Administration of Affairs Concerning Experimental Animals of China. The study is also reported in accordance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chang, L., Zheng, Y., Li, S. et al. Identification of genomic characteristics and selective signals in Guizhou black goat. BMC Genomics 25, 164 (2024). https://doi.org/10.1186/s12864-023-09954-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09954-6