
Abstract
Background
Poplar (Populus cathayana)and willow (Salix rehderiana) are important fast-growing trees in China. Grafting plays an important role in improving plant stress resistance and construction of ornamental plants. It is found that willow scions grafted onto poplar rootstocks can form ornamental plants. However, this grafted combination has a low survival rate. Many studies have reported that the xyloglucan endotransglucosylase/hydrolase (XTH) family plays an important role in the healing process of grafts.
Results
A total of 38 PtrXTHs and 32 SpuXTHs were identified in poplar and willow respectively, and were classified into three subfamilies. Tandem duplication was the main reason for the expansion of the PtrXTHs. Grafting treatment and Quantitative real time PCR (RT-qPCR) analysis revealed that five XTH genes differentially expressed between self-grafted and reciprocal grafted combinations. Specifically, the high expression levels of SrXTH16, SrXTH17, SrXTH25, PcXTH22 and PcXTH17 may contribute to the high survival rate of the grafted combination with willow scion and poplar rootstock. Subcellular localization identified that the SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 proteins were located on the cell walls. Transcription factors (NAC, MYB and DOF) may regulate the five XTH genes.
Conclusions
This study provides a new understanding of the roles of PcXTH and SrXTH genes and their roles in grafting. Our results will give some hints to explore the molecular mechanisms of PcXTH and SrXTH genes involved in grafting in the future.
Similar content being viewed by others


Background
Salicaceae plants have about 50 genera, e.g., Populus, Chosenia and Salix, with ~ 1000 species all over the world [1]. P. cathayana and S. rehderiana are fast-growing native species and have important ecological value in China [2,3,4]. As for their strong adaptability and cold resistance, the two species have been the most intensive plantation species, and widely distribute in the eastern Tibetan Plateau [5, 6]. Grafting can produce plants with good characteristics, and is widely used in horticulture, agriculture and forestry. Compared to intra-specific grafted plants, inter-specific grafted plants are usually more difficult to obtain and show greater heterosis [7]. Recently, we have successfully performed the inter-generic grafting between Populus and Salix, in which the combination of willow as scion and poplar as rootstock is the highest survival rate [8]. However, it is still unknown what the molecular mechanisms of the different graft survival rates.
In recent years, research on the grafted healing site has become a research hotspot. The graft healing process has been identified as five stages, including cambial tissue arrangement, isolation layer appearance, callus formation, cambium restoration, and connective tissue connection [9]. New vascular formation is the key point for successful grafting [9, 10]. The WUSCHEL-related homeobox 4 (WOX4), wound induced dedifferentiation 1/2/3 (WIND1/2/3), and WOX13 have been shown to play important roles during vascular formation process in the grafted plants [11]. RNA-seq analyses also indicated that cell wall-related genes played significant roles in promoting plant grafting healing [12, 29] (Fig. 1A, B). The SpuXTH32 was distinctively distributed on one chromosome segment due to the incomplete genome assembly. The rest of the SpuXTHs were irregularly distributed on the chromosomes except Chr12, Chr15W, Chr15Z, Chr16 and Chr17. PtrXTHs were sporadically located on the chromosomes except Chr12, Chr15 and Chr17. The maximum number of XTH genes on one chromosome was higher in poplar than in willow, and the minimum number was equal.

The collinearity analysis of Populus, Salix and Arabidopsis. Firstly, the collinearity analysis within species were conducted, including Salix (A) and Populus (B). Then the collinearity analysis among species were conducted, including Populus and Arabidopsis, Salix and Arabidopsis, and Populus and Salix (C). The gray lines in the background represent collinear blocks between genomes of species, while the black, orange, blue and red lines highlight the syntenic XTH gene pairs
The gene duplication, a force of gene expansion and evolution, is usually be divided into two situations, the tandem duplication and the segmental duplication. Tandem duplication is usually found in gene clusters to uncover the expansion of gene families [30]. We found three gene clusters in Salix, located on chromosomes 02, 05, and 11, respectively, including six XTH genes (SpuXTH13-14, SpuXTH11-12, SpuXTH22-23) (Fig. 1A). Four gene clusters, PtrXTH3-4, PtrXTH11-12, PtrXTH24-25 and PtrXTH34-37, were respectively located on Chr02, Chr05, Chr11 and Chr18 in Populus (Fig. 1B). Inter-species collinearity analysis revealed that there were 12 segmental duplication events which involved 24 genes in poplar and 7 which contained 14 genes in willow. These gene pairs were used to calculate Ka (synonymous substitution)/Ks (non-synonymous substitution) values to assess the evolutionary selective pressure on XTH genes (Table S2). Results showed that 18 XTH gene pairs were detected with the Ka/Ks < 1, which suggested that these genes have suffered strong purifying selection. The sequences of PtrXTH12 and PtrXTH24 are highly diverge and have a long evolutionary distance, thus the Ka/Ks value is null.
To further analyze the evolution of XTH genes in plants, we conducted collinearity analysis in Salix, Populus and Arabidopsis (Fig. 1C). Syntenic maps revealed that there were 50 pairs of homologous genes between Salix and Populus, 24 pairs of homologous genes between Salix and Arabidopsis, and 26 pairs of homologous genes between Populus and Arabidopsis. Thus, there is a closer homology of XTH genes in Salix and Populus than those in Arabidopsis with Salix or Populus.
Phylogenetic analysis and classification of XTH proteins
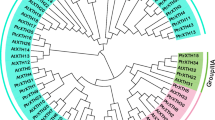
To elucidate the evolutionary relationship, we used multiple sequence alignment to analyze the 103 full-length XTH protein sequences from Salix, Populus and Arabidopsis. The neighbor-joining method was used to construct the phylogenetic tree (Fig. 2). The phylogenetic tree consists of three predominant branches. 73 proteins are classified into the largest I/II group. Within this group, 23 XTH proteins belonged to willow and 28 belonged to poplar. The III group can be subdivided into III-A and III-B groups. The phylogenetic analysis indicated that SpuXTH14, SpuXTH18, PtrXTH14, PtrXTH19 and PtrXTH32 belonged to the III-A group, in which these genes had XEH activity. SpuXTH2, SpuXTH7, SpuXTH17, SpuXTH20, SpuXTH21, SpuXTH26, PtrXTH2, PtrXTH7, PtrXTH18, PtrXTH21, PtrXTH22 and PtrXTH28 were grouped into III-B group, which mainly exhibited XET activity. The ancestral group had the fewest XTH proteins, including AtXTH1, AtXTH2, AtXTH3, AtXTH11, SpuXTH6 and PtrXTH6.

Evolutionary and phylogenetic analysis of the XTH genes family in Populus, Salix and Arabidopsis. The bootstrap values were listed in the evolutionary tree. Different shapes represented different species. The red circle represented Populus. The red box represented Salix. The red five-pointed star represented Arabidopsis. The colors of the outer ring and branches indicated different group
Gene structure and conserved motifs of XTH
To gain further insight into the structural diversity of the XTH genes in poplar and willow, the structure of the 32 SpuXTHs and 38 PtrXTHs was identified using TBtools software based on genomic sequences (Fig. 3). The number of exons varied from one to five in willow and from one to four in poplar (Fig. 3). SpuXTH20 and PtrXTH21 have no intron and untranslated region (UTR). SpuXTH3, SpuXTH10, SpuXTH23, SpuXTH29 and PtrXTH31 have no UTR either.

Phylogenetic and motifs analysis of PtrXTH and SpuXTH proteins, and gene structure and conserved domain structure analysis of PtrXTH and SpuXTH genes. A Phylogenetic tree and protein motifs were showed using full-length amino acid sequences from SpuXTHs. Blue represents I/II group, pink represents ancestral group, red represents III-A group and orange represents III-B group. Exon–intron structures and conserved domains were analyzed using gene sequences of SpuXTHs. B Phylogenetic tree and motifs were showed using full-length amino acid sequences from PtrXTHs. Blue represents I/II group, pink represents ancestral group, red represents III-A group and orange represents III-B group. Exon–intron structures and conserved domains were analyzed using gene sequences of PtrXTH genes
Glyco_hydro_16 and XET_C are conserved domains located on the XTH (Fig. 3). Most of the SpuXTH and PtrXTH proteins contained a putative signal peptide, which was a short peptide that is the key for translocation to the cellular membrane or extracellular space [31]. Additionally, the low_complexity_region, which could expand protein sequence space was identified in 24 XTH genes in willow and 15 XTH genes in poplar [32]. The transmembrane_domain, comprising a stretch of 17-25 hydrophobic amino acid residues that are structured as an α-helix, was found in 18 SpuXTH genes and 2 PtrXTH genes [33].
The conserved motifs were predicted as well (Fig. 3). Almost all XTHs in the same group shared common motifs. Motifs 1-3 and motif 5 were highly conserved in all SpuXTHs, while motifs 1-2, and motif 5 were highly conserved in all PtrXTHs. They were possibly composed of specific conserved domains Glyco_hydro_16 and XET_C. The proteins had six to ten motifs in willow and poplar. Some I/II group proteins in willow had motif 6 and motif 9, while other proteins in I/II group did not have. The III-B group proteins had the same motifs, except for SpuXTH17. The III-A group was consisted of SpuXTH14 and SpuXTH18, which had XEH activity, probably because they lacked motif 6 and motif 14. The motifs of PtrXTHs in the I/II group were multiple, indicating that these proteins have different functions. Except for PtrXTH23 and PtrXTH31, the rest of the genes in I/II group had motifs 1-5. Additionally, proteins in I/II group either had motif 6 or motif 11. The proteins of poplar in III-B group shared the same motifs, which indicated that the encoded proteins had similar structure and function. The PtrXTH proteins in the III-A group had a special motif 9 that wasn’t identified in other groups, which might be the reason why they had XEH activity.
Structure-based sequence alignment and prediction of protein tertiary structure
The proportion of XTHs secondary structure was predicted (Table S3). The random coil accounted for ~ 30% and the extended strand accounted for ~ 50% of the secondary structure in both willow and poplar. The β-turn had the lowest proportion. The schematic of the secondary structures showed that SpuXTH and PtrXTH proteins contained the conserved E × D × E × domain, acting an active site (Fig. 4). Except for the ancestral and III-A group members, I/II and III-B group had an N-glycosylation domain. Additionally, there are two conserved domains after the conserved E × D × E × domain, loop 2 and loop 3. Loop 2 in I/II, III-B and ancestral group members was shorter than that in III-A group. A conserved WAT × GG sequence (loop 3) of I/II, III-B and ancestral group members or a conserved SWATE × sequence (loop 3) of III-A group members was located in the C-terminal region. The C-terminal region of XTH proteins often contains highly conserved cysteines, which can form disulfide bonds and contribute to the stability of XTHs' protein structure [34].

Structure-based sequence alignment of SpuXTH and PtrXTH proteins. A The SpuXTH proteins of Group I/II, III-B and ancestral group. B The SpuXTH proteins of III-A group. C The tertiary structure of SpuXTH6, SpuXTH14 and SpuXTH25. D The PtrXTH proteins of Group I/II, III-B and ancestral group. E The PtrXTH proteins of III-A group. F The tertiary structure of PtrXTH6, PtrXTH19 and PtrXTH26. The secondary structures of α-helices (spiral), β-sheets (arrows) and N-glycosylation site (*) are indicated. Leu is short for leucine. Phe is short for phenylalanine. Ile is short for isoleucine
Six XTH proteins with different amino acids at the activation sites, randomly selected from poplar and willow, were predicted using three-dimensional structure simulations (Fig. 4C, F). The homology modeling method was used to predict the tertiary structure of unknown proteins using homologous protein with known tertiary structure in the database. The results revealed that the six protein models consisted of large β-sheets arranged in a sandwich manner. Differences in amino acids at the activation sites led to subtle differences in tertiary structure and function between the proteins. These proteins are ready for further study.
Analysis of cis-elements in XTHs
The 2000 bp upstream sequence of coding sequence (CDS) was extracted to predict cis-elements in SpuXTH and PtrXTH genes (Fig. 5). Most of the cis-elements, e.g., I-box, TCT-motif, GT1-motif, GA-motif, TCCC-motif, AE-box, AT1-motif, ATC-motif, G-box, GATA-motif, box 4 and MRE, were classified as light-responsive elements. Circadian rhythm control, meristem expression and zein metabolism regulation elements were related to growth and development. Anaerobic induction, defense and stress response, drought induction and low temperature induction elements were related to abiotic stress. Salicylic acid response, methyl jasmonate, abscisic acid, gibberellin and auxin response elements were included in the hormone related elements. Among them, the abundances of auxin and methyl jasmonate elements were contained in XTH genes, and these XTH genes may be regulated during grafting process.

Analysis of cis-elements in SpuXTH and PtrXTH promoters. A Number of distinct cis-elements in each SpuXTH genes. B Number of distinct cis-elements in each PtrXTH genes
Expression pattern of XTH genes in grafted plants
We analyzed the expression pattern of XTH genes in the four grafted combination: P. cathayana grafted onto P. cathayana (P/P), P. cathayana grafted onto S. rehderiana (P/S), S. rehderiana grafted onto P. cathayana (S/P) and S. rehderiana grafted onto S. rehderiana (S/S) (Fig. 6). In the self-grafted combinations, XTH genes were highly expressed after three and ten days of grafting. Two SpuXTH genes (SpuXTH23 and SpuXTH33) and four PtrXTH (PtrXTH6, PtrXTH21, PtrXTH24 and PtrXTH25) genes didn’t express in four grafted plants. The expression level of most XTH genes is higher in self-grafted combinations than in reciprocal-grafted combinations. At three, ten and twenty-eight days, the PtrXTH genes were highly expressed in S/P and the SpuXTH genes were highly expressed in P/S.

Morphology of four grafted combinations (P/P, P/S, S/S and S/P) and expression patterns of PcXTH and SrXTH genes. A Four grafting combinations of poplar and willow at 3, 10, and 28 days after grafting. B Heat map of the expression levels of SrXTH genes on the third, tenth and twenty-eighth days after grafting in grafted combinations S/S, P/S and S/P based on FPKM values. Heat map of the expression levels of PcXTH genes on the third, tenth and twenty-eighth days after grafting in grafted combinations P/P, P/S and S/P based on FPKM values
The quantitative real time PCR (RT-qPCR) results showed that compared with S/S, SrXTH16 and SrXTH17 had lower expression in P/S and S/P, while SrXTH25 was highly expressed in P/S and S/P (Fig. 7A). Compared with the P/P, PcXTH17 and PcXTH22 had low expression in P/S and S/P during graft healing process (Fig. 7B). To gain their molecular functions, the SrXTH16-GFP, SrXTH17-GFP, SrXTH25-GFP, PcXTH17-GFP and PcXTH22-GFP plasmids were transferred into Agrobacterium. The subcellular results showed that the SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 proteins were located on the cell wall (Fig. 8).

Expression profiles of SrXTH and PcXTH genes in different grafted combinations after three, ten and twenty-eight days. Error bars on the graph indicate the mean standard deviation for each triplicate treatment

Subcellular localization of the SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 proteins. pCAMBIA2300-GFP (empty vector), pCAMBIA2300-SrXTH16, pCAMBIA2300-SrXTH17, pCAMBIA2300-SrXTH25, pCAMBIA2300-PcXTH17 and pCAMBIA2300-PcXTH22 were transiently expressed in onion epidermal cell layers. Scale bar = 50.0 μm
Prediction of transcription factors that regulated XTH genes
A total of 323 transcription factors were predicted to regulate SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 (Fig. 9A and B). They were 13 of SrXTH16, 125 of SrXTH17, 19 of SrXTH25, 60 of PcXTH17 and 106 of PcXTH22. All these XTH genes were predicted to be regulated by members of the NAC, DOF (DNA binding with one finger), HD-Zip (homeodomain-leucine zipper) and MYB (v-myb avian myeloblastosis viral oncogene homology) transcription factor families, such as MYB6 (Potri.004G088100), DOF1.4 (Potri.011G055600), HAT4 (Potri.014G045100) and NAC017 (Potri.002G061300). PcXTH17, SrXTH17 and SrXTH25 were regulated by ethylene responsive factor (ERF) transcription factor family, e.g., ERF016 (Potri.018G038100), ERF106 (Sapur.001G064100) and DREB3 (Sapur.003G026800). PcXTH17 and SrXTH17 genes were regulated by the auxin response factors (ARF), e.g., ARF19 (Sapur.006G114200) and ARF2A (Potri.015G105300). Then we performed Gene Ontology (GO) enrichment analysis on predicted transcription factors (Fig. 9C). In addition to the functions related to gene regulation, 208 transcription factors were also involved in cellular biosynthetic process and cellular metabolic biosynthesis process. These indicated that these XTH genes may participate in the graft healing process by regulating cell division and growth.

Predicted transcription factors that regulate XTH genes and GO analysis of the selected transcription factors. A and (B) There are 323 pairs of interacting proteins for 5 XTH genes. The red circle represents the PcXTH or SrXTH genes. The blue circle represents the transcriptional factor in each clade. The thickness of the green line indicates the reliability of the predicted results. The line is thicker, the result is more reliable
Discussion
Poplar grafted to willow has a higher successful rate than willow grafted to poplar [8]. However, the molecular mechanism is not clear. XTH proteins are a kind of xyloglucan transferases/hydrolases involved in the regulation of cell wall relaxation. Due to their roles in cell wall modification, we believe that the XTH gene may have important effects on the grafted healing.
38 and 33 XTH genes were respectively identified in poplar and willow. They were divided into four groups: I/II, III-A, III-B and ancestral group. The XET proteins belonging to the I/II and III-B groups have a conserved N-linked glycosylation site, which is thought to be important for protein stability [19, 29, 35]. N-glycosylation domain was absent almost in all III-A and ancestral group members, which was consistent with our results [36, 37]. Therefore, the XTH proteins of I/II and III-B group have higher protein stability than that of III-A and ancestral group. XTH proteins belonging to I/II, III-B and ancestral group have XET activity, while XTH proteins belonging to III-A group have XEH activity [38]. The length of loop 2 was the key point that determined the XET or XEH activity of XTH proteins. The loop 2 of I/II, III-B and ancestral group is shorter than that of III-A group proteins [29, 37]. XET proteins specifically recognizes xyloglucan and forms new xyloglucan chains to expand call walls [39]. Previous research found that high expression of β-1,4-glucanase make tobacco successfully graft with 73 species of plants [28]. Evidence shows that it plays an important role in the grafting process of tobacco by facilitating cell wall reconstruction near the graft interface [28]. Because XETs can expand cell wall to promote cell wall construction, we believe that XET enzymes play an important role in grafting process.
Heatmap results showed that some genes didn’t express and some genes highly expressed during graft healing process. Phylogenetic analysis showed that the SpuXTH6 and PtrXTH6 were assigned to the ancestral group. A recent study reported that genes in the ancestral group were not the ancestors of the other group genes, but diverged after other groups, possibly introducing some new roles in flowering plants [26]. They were predominantly or exclusively expressed in reproductive tissues of flowering plants but not found in any lower plants [26]. This may be the reason that the expression levels of SrXTH6 and PcXTH6 were not detected in stem tissue. SpuXTH22, SpuXTH23, PtrXTH24 and PtrXTH25 belonged to I/II group. AtXTH15, which was homologous to them, was related to shade and mainly expressed in seeds [40]. PtrXTH21 belonged to III-B group. AtXTH27, which was homologous to it, regulated tracheary element development [41]. The SpuXTH16, SpuXTH25 and PtrXTH17 belonged to I/II group, while SpuXTH17 and PtrXTH22 belonged to III-B group. They highly expressed in the stem. AtXTH9, homologous to SpuXTH16, SpuXTH25 and PtrXTH17, regulated secondary call wall formation [42]. The subcellular localization results indicated that SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 proteins were located on cell walls.
Many genes encode cell wall modification enzymes play important roles during graft healing process [11]. Genes encoded cell wall-plasma membrane linker proteins and cell wall LRR proteins and genes related to phloem development and cellulose biosynthesis were up-regulated at 3-28 days after grape self-grafted plants [12]. AtXTH28 highly expressed after Arabidopsis and tobacco grafted combination [28]. Our results also found that SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 highly expressed at 3-28 days after poplar and willow self-grafted combinations. The up-regulated XTH genes may lead to the accumulation of xyloglucan in the cell wall of tomato graft interface to promote cell adhesion [14]. Cell wall modification related genes actively expressed, causing significant changes in the cell wall to promote the formation of secondary plasmodesmata at the grafted junction [43]. It provides energy for rootstocks and promotes the material exchange between rootstocks and scions [43]. These are key points to improve the survival rate of grafting [44]. The high survival rate of reciprocal grafted plants is also related to the positive expression of cell wall related genes [45, 46]. Our results found that the expression levels of PcXTH22 and PcXTH17 in the low survival rate grafted combination S/P were higher than those in the high survival rate grafted combination, which may play an important regulatory role in the healing process of S/P grafting. SrXTH16, SrXTH17 and SrXTH25 have higher expression levels in P/S than in S/P, which may be important genes affecting the survival rate of reciprocal graft between poplar and willow.
The expression levels of XTH genes are regulated by transcription factors [27, 47]. They binding with cis-elements of genes is one of the important mechanisms regulating expressions of genes [48,49,50]. Based on this fact and gene co-expression network, we predicted transcription factors that regulated five XTH genes: SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22. Results showed that all five XTH genes were regulated by NAC, MYB and DOF transcription factors. NAC transcription factors, e.g., NST3/ANAC012, NST1/ANAC043, PtrWND2 and PtrWND6, activate plant secondary cell walls growth program resulting in ectopic deposition of secondary cell walls [47, 51]. The AtXTH10 was directly regulated by NST3 and VND7 [47]. In our study, PcXTH22 was regulated by PtrNAC017(Potri.002G061300). Previous study found that ANAC071, homologous with PtrNAC071, bound to the AtXTH19 and AtXTH20 promoters to induce their expression in the distal part of an incised stem and their involvement in cell proliferation in the tissue reunion process [27]. MYB transcription factors, e.g., MYB46, MYB83 and MYB103 were regulated by secondary wall NAC transcription factors to participate in the biosynthesis of cell walls [47, 52]. In self-grafted pepper plants, MYB86 was predicted to regulate XTH22 and XTH38 [53]. Our study found that PcMYB61 (Sapur.005G000800) regulated SrXTH25. AtMYB46, which was homologous with PtrMYB61, can activate the synthesis of cellulose and hemicellulose [54]. DOF transcription factors participates in regulating tissue differentiation and vascular system development [50]. Overexpression of AtDOF5.4 in Arabidopsis plants exhibited dwarfing phenotypes [55]. AtDOF5.8 was thought to play important role in the early stages of primitive meristem cell formation and vascular system formation [50]. In our results, PcXTH17 was regulated by PcDOF1.2 (Potri.002G070700). The AtDOF2.1 transcription factor, which is homologous to DOF1.2, was induced after injury and involved in cell wall modification [56]. Existing grafting experiments showed that four DOF transcription factors (HCA2, TMO6, DOF2.1, and DOF6) mutant plants inhibited phloem reconnection after grafting, while overexpressing plants accelerated phloem reconnection after grafting [56]. Therefore, we thought that the NAC, MYB, and DOF family transcription factors may participate in the grafting healing process by regulating the XTH gene.
Conclusions
In this study, 38 PtrXTH genes and 32 SpuXTH genes were identified, and they were divided into three subfamilies. Tandem duplication contributed most to the expansion of the PtrXTH family. Five XTH genes (SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22) are thought to play important roles in Salicaceae grafting and are regulated by some transcription factors (NAC, MYB and DOF) during graft healing process. In summary, this study improved our understanding of the PcXTH and SrXTH gene families and lay the foundation for further exploration on grafting in poplar and willow.
Methods
Identification of XTH family genes
Gene, protein and gene annotation files of P. trichocarpa and S. sinopurpurea were downloaded from the Phytozome website (https://phytozome-next.jgi.doe.gov/). Meanwhile, the protein and gene sequences of 33 XTH family genes identified in Arabidopsis were also downloaded from Phytozome [57].
Two methods were used to identify XTH family genes in Salicaceae. To obtain profiles of XTH protein domains, the query numbers PF00722 and PF06955 found in literatures were used to search in the pfam-A database (http://pfam.xfam.org/), which was derived from the pfam website [24]. Using the query results, the Simple Hidden Markov Model (HMM) Search function in TBtools software could retrieve all potential XTH family members from whole genome sequences in P. trichocarpa and S. sinopurpurea [58]. The threshold used for identifying the XTHs domains was 1E-5. Another way was to perform a two- way blast alignment using the Two Sequence Files tool in TBtools in combination with the protein sequences of Arabidopsis XTH family genes and whole genome sequences of P. trichocarpa and S. sinopurpurea to identify all potential XTH family genes in Salicaceae [58]. To further screen the XTHs, the Batch SMART function of TBtools software was used to visualize the protein domains of XTHs obtained separately by the two methods [58, 59]. After removing the domain mismatch and deficiency, 32 SpuXTHs and 38 PtrXTHs were identified (Table S4).
Physical and chemical analysis and the secondary and tertiary structure of proteins
The length, molecular weight, theoretical isoelectric point, instability index, aliphatic index and grand average of hydropathicity of XTH proteins were characterized by using the online tool ExPASy ProtParam (https://web.expasy.org/protparam/) [60]. The secondary structure of XTH proteins was predicted by the online tool SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa%20_sopma.html). Finally, the online tool SWISS-Model Interactive Workespace (https://swissmodel.expasy.org/interactive) was used to predict the tertiary structure of XTH proteins by homolog modeling method.
Analysis of gene structure, protein conserved motif and protein conserved domain
The conserved motif profiles of the proteins were predicted by MEME Suite 5.4.1 (https://meme-suite.org/meme/tools/meme) using default parameters, and the motif number was set to 15 for poplar and willow [61]. The profiles of conserved protein domains were predicted and listed by Batch SMART tool of TBtools software. Then, the evolutionary tree, gene structure, domain, and protein motif were draw using Gene Structure View tool of TBtools software.
The collinearity analysis, structural-based sequence alignment and phylogenetic analysis
The One step MCScan X tool in TBtools software was used for the collinearity analysis [58]. The secondary structures of PttXET16-34 (PDB id:1UN1) and TmNXG1 (PDB id: 2UWA) were downloaded from the PDB database (https://www.rcsb.org/) and used as a comparison standard [62]. The Align by Clustal W tool with default parameters in MEGA11.0 was used for multiple sequence alignment with downloaded data and protein sequences of poplar and willow [63, 64]. The multiple sequence alignment results were analyzed and visualized using ESPript 3.0 version (https://espript.ibcp.fr/ESPript/ESPript/index.php) to predict the secondary structures of SpuXTHs and PtrXTHs. The phylogenetic tree was constructed by neighbor joining method using multiple sequence alignment. The bootstrap value was set at 1000. The evolutionary tree was modified by Evolview3.0 online software [64].
Calculation of Ka/Ks
The Ka, Ks and their ratio of XTH pairs were calculated using the Simple Ka/Ks Calculator function of TBtools software [58]. Ks is the calculated value of synonymous mutation frequency and Ka is the calculated value of non-synonymous mutation frequency [65]. The ratio of Ka/Ks can be used to estimate the selection pressure. If the ratio is greater than 1, it means positive selection [66]. And if the ratio is less than 1, it indicates purity selection [66]. If the ratio is the equal to 1, it means neutral selection [66].
Analysis of cis-acting elements
Firstly, using TBtools’ GTF/GFF3 Sequences extractor function to extract 2000 bp upstream of the CDS sequences of XTH family genes, then predicting cis-acting elements using Plant Care database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). The results were visualized by HeatMap tool of TBtools software.
Plant materials and transcriptome analysis
Four grafting combinations (scion/rootstock: P/P, S/S, P/S, S/P) were made using P. cathayana (P) and S. rehderiana (S). After three, ten and twenty-eight days of grafting, callus was collected from the grafted healing sites (five centimeters each from the contact area of the scion and rootstock). Fifteen grafted plants were performed at each time point, and nine grafted plants were randomly selected and divided into three groups for callus collection. Samples collected at three, ten and twenty-eight days were placed into liquid nitrogen. Total RNA was extracted using Trizol reagent kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. The Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) was used to assess RNA quality and the RNase free agarose gel electrophoresis was used to verify the results. After total RNA extraction, eukaryotic mRNA was enriched by Oligo(dT) beads, and prokaryotic mRNA was enriched by removing rRNA by Ribo-ZeroTM Magnetic Kit (Epicentre, Madison, WI, USA). Then the enriched mRNA was fragmented into short fragments using fragmentation buffer and reverse transcribed into cDNA by using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB #7530, New England Biolabs, Ipswich, MA, USA). The purified double-stranded cDNA fragments were end repaired, A base added, and ligated to Illumina sequencing adapters. The ligation reaction was purified with the AMPure XP Beads (1.0X), and amplified by polymerase chain reaction (PCR). The cDNA library was sequenced using Illumina Novaseq6000 by Gene Denovo Biotechnology Co. (Guangzhou, China).The paired-end clean reads were mapped to the reference genomes (P. trichocarpa and S. purpurea genomes). For each transcription region, a FPKM (fragment per kilobase of transcript per million mapped reads) value was calculated to quantify its expression abundance and variations.
Experimental validation of XTH transcript levels by RT-qPCR analysis
After three, ten and twenty-eight days of grafting, the grafted healing sites were collected and frozen in liquid nitrogen. All samples were then stored at -80 °C. Total RNA was extracted using a RNeasy kit (Tiangen, Biotech) according to the instructions. Total RNA (1 μg) was treated with a FastQuant RT kit (with gDNase; Yeasen, https://www.yeasen.com/) to get reverse transcript. 18S ribosomal RNA (rRNA) was used as the reference gene [67]. The primer sequences presented in Table S5 were designed by Primer Premier 6.0 and blasted in NCBI. RT-qPCR analysis was performed using LineGene 9600 Plus platform (Bio-er, Hangzhou). The transcript levels were analyzed using the 2−ΔΔCt method and means ± standard errors (SE) [68].
Subcellular localization of the XTH proteins
The CDS of SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22 were amplified by PCR. Link PCAMBIA2300-GFP vector was linked to the amplification products by homologous recombination method. The linked products were first introduced into DH5α-competent cells to screen for plasmids with correct sequences, which were then introduced into Agrobacterium tumefaciens GV3101 strain for transient expression in the epidermal cell layer of onion bulbs that incubated on medium for 1 day in advance. Infected onion epidermal cell layers were maintained at 25 °C in the dark and harvested for fluorescence examination 48 h after infection. GFP signals in the transiently infected onion epidermis were observed using a Leica DM4 B upright fluorescence microscope (Leica, Germany). A 30% sucrose solution was used to separate onion cytoplasmic walls. The relevant primers are listed in supplementary Table S5.
Prediction of transcription factors that regulated XTH genes
The potential transcription factors that regulated five XTH genes (SrXTH16, SrXTH17, SrXTH25, PcXTH17 and PcXTH22) were predicted using the transcription factor targeting analysis tool on the GENE DENOVO cloud platform (https://www.omicsmart.com/index.php). Pearson correlation analysis was performed using the expression levels of XTH genes and transcription factors in R software using the readxl (version 1.4.3) and stringr (version 1.5.0) packages, and transcription factors with correlation coefficients greater than or equal to 0.8 were screened for plotting on the GENE DENOVO cloud platform (Table S6). In addition, GO analysis of the selected transcription factors was performed on the Omicsmart online platform ((https://www.omicsmart.com/). The P value <0.01.
Availability of data and materials
The data reported in this paper have been deposited in the OMIX, China National Center for Bioinformation / Bei**g Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn/omix/release/OMIX004709).
Abbreviations
- ARF:
-
Auxin response factor
- CDS:
-
Coding sequence
- DOF:
-
DNA binding with one finger
- GH16:
-
Glycoside hydrolase family
- GO:
-
Gene Ontology
- HD-Zip:
-
Homeodomain-leucine zipper
- HMM:
-
Hidden Markov Models
- JA:
-
Jasmonic acid
- Ka:
-
Synonymous substitution
- Ks:
-
Non-synonymous substitution
- MeJA:
-
Methyl jasmonate
- MW:
-
Molecular weight
- MYB:
-
V-myb avian myeloblastosis viral oncogene homology
- NAC:
-
NAC domain containing protein
- pI:
-
Isoelectric point
- UTRs:
-
Untranslated regions
- WIND1/2/3:
-
Wound induced dedifferentiation 1/2/3
- WOX4:
-
WUSCHEL-related homeobox 4
- XEH:
-
Xyloglucan endoglucosidase
- XET:
-
Xyloglucan endohydrolase
- XTH:
-
Xyloglucan endotransglucosylase/hydrolase
References
Gulyaev S, Cai XJ, Guo FY, Kikuchi S, Applequist WL, Zhang ZX, et al. The phylogeny of Salix revealed by whole genome re-sequencing suggests different sex-determination systems in major groups of the genus. Ann Bot. 2022;129(4):485–98.
Zhou J, Zhuo R, Liu M, Qiao G, Jiang J, Li H, et al. Identification and Characterization of Novel MicroRNAs from Populus cathayana Rehd. Plant Mol Biol Report. 2010;29(1):242–51.
Han YY, Luo X, Zhang WR, Xu TY, Tang M. Arbuscular mycorrhizal fungi enhanced drought resistance of Populus cathayana by regulating the 14-3-3 family protein genes. Microbiology Spectrum. 2022;10(3):e0245621.
Ran X, Huang X, Wang X, Liang H, Wang Y, Li J, et al. Ion absorption, distribution and salt tolerance threshold of three willow species under salt stress. Front Plant Sci. 2022;13:969896.
Chen L, Mi J, Hao L, He F, Yang H, Wan X, et al. Effects of simulated nitrogen deposition on the ecophysiological responses of Populus bei**gensis and P. cathayana under intra- and interspecific competition. Plant and Soil. 2022;481(1–2):127–46.
Cai Z, Fu M, Yao Y, Chen Y, Song H, Zhang S. Differences in phytohormone and flavonoid metabolism explain the sex differences in responses of Salix rehderiana to drought and nitrogen deposition. Plant J. 2023;114(3):534–53.
Han Q, Song H, Yang C, Zhang S, Korpelainen H, Li C. Integrated DNA methylation, transcriptome and physiological analyses reveal new insights into superiority of poplars formed by interspecific grafting. Tree Physiol. 2022;42(7):1481–500.
Han Q, Song H, Tang D, Zhang S. Physiological responses of interspecific grafted seedlings of Salix sichuanensis and Populus cathayensis to nitrogen deficiency (in chinese). Journal of Applied Ecology 2017;28(12):3833–40.
Baron D, Esteves Amaro AC, Pina A, Ferreira G. An overview of grafting re-establishment in woody fruit species. Sci Hortic-Amsterdam. 2019;243:84–91.
Wang J, Jiang L, Wu R. Plant grafting: how genetic exchange promotes vascular reconnection. New Phytol. 2017;214(1):56–65.
Ikeuchi M, Iwase A, Ito T, Tanaka H, Favero DS, Kawamura A, et al. Wound-inducible WUSCHEL-RELATED HOMEOBOX 13 is required for callus growth and organ reconnection. Plant Physiol. 2022;188(1):425–41.
Cookson SJ, Clemente Moreno MJ, Hevin C, Nyamba Mendome LZ, Delrot S, Trossat-Magnin C, et al. Graft union formation in grapevine induces transcriptional changes related to cell wall modification, wounding, hormone signalling, and secondary metabolism. J Exp Bot. 2013;64(10):2997–3008.
**e L, Dong C, Shang Q. Gene co-expression network analysis reveals pathways associated with graft healing by asymmetric profiling in tomato. BMC Plant Biol. 2019;19(1):373.
Frey C, Manga-Robles A, Acebes JL, Encina A. The graft framework: Quantitative changes in cell wall matrix polysaccharides throughout the tomato graft union formation. Carbohydr Polym. 2022;276:118781.
Melnyk CW. Plant grafting: insights into tissue regeneration. Regeneration (Oxf). 2017;4(1):3–14.
Cheng Z, Zhang X, Yao W, Gao Y, Zhao K, Guo Q, et al. Genome-wide identification and expression analysis of the xyloglucan endotransglucosylase/hydrolase gene family in poplar. BMC Genomics. 2021;22(1):804.
Zhu J, Tang G, Xu P, Li G, Ma C, Li P, et al. Genome-wide identification of xyloglucan endotransglucosylase/hydrolase gene family members in peanut and their expression profiles during seed germination. PeerJ. 2022;10:e13428.
Ishida K, Yokoyama R. Reconsidering the function of the xyloglucan endotransglucosylase/hydrolase family. J Plant Res. 2022;135(2):145–56.
Johansson P, Brumer H 3rd, Baumann MJ, Kallas AM, Henriksson H, Denman SE, et al. Crystal structures of a poplar xyloglucan endotransglycosylase reveal details of transglycosylation acceptor binding. Plant Cell. 2004;16(4):874–86.
Atkinson RG, Johnston SL, Yauk Y-K, Sharma NN, Schröder R. Analysis of xyloglucan endotransglucosylase/hydrolase (XTH) gene families in kiwifruit and apple. Postharvest Biol Technol. 2009;51(2):149–57.
Wu D, Liu A, Qu X, Liang J, Song M. Genome-wide identification, and phylogenetic and expression profiling analyses, of XTH gene families in Brassica rapa L. and Brassica oleracea L. BMC Genomics. 2020;21(1):782.
Saab IN, Sachs MM. A flooding-induced xyloglucan endo-transglycosylase in maize is responsive to ethylene and associated with aerenchyma. Plant Physiol. 1996;111(2):110.
Uozu S, Tanaka-Weguchi M, Kitano H, Hattori K, Matsuoka M. Characterization of XET-related genes of rice. Plant Physiol. 2000;122(3):853–9.
Wang M, Xu Z, Ding A, Kong Y. Genome-wide identification and expression profiling analysis of the xyloglucan endotransglucosylase/hydrolase gene family in tobacco (Nicotiana tabacum L.). Genes (Basel). 2018;9(6):273.
Yang Z, Zhang R, Zhou Z. The XTH gene family in Schima superba: Genome-wide identification, expression profiles, and functional interaction network analysis. Front Plant Sci. 2022;13:911761.
Seven M, Derman UC, Harvey AJ. Enzymatic characterization of ancestral/group-IV clade xyloglucan endotransglycosylase/hydrolase enzymes reveals broad substrate specificities. Plant J. 2021;106(6):1660–73.
Pitaksaringkarn W, Matsuoka K, Asahina M, Miura K, Sage-Ono K, Ono M, et al. XTH20 and XTH19 regulated by ANAC071 under auxin flow are involved in cell proliferation in incised Arabidopsis inflorescence stems. Plant J. 2014;80(4):604–14.
Notaguchi M, Kurotani K, Sato Y, Tabata R, Kawakatsu Y, Okayasu K, et al. cell-cell adhesion in plant grafting is facilitated by β-1,4-glucanases. Science. 2020;369(6504):698.
Wu J, Zong Y, Tu Z, Yang L, Li W, Cui Z, et al. Genome-wide identification of XTH genes in Liriodendron chinense and functional characterization of LcXTH21. Front Plant Sci. 2022;13:1014339.
Huang YL, Zhang LK, Zhang K, Chen SM, Hu JB, Cheng F. The impact of tandem duplication on gene evolution in Solanaceae species. J Integr Agric. 2022;21(4):1004–14.
Holec PV, Camacho KV, Breuckman KC, Mou J, Birnbaum ME. Proteome-scale screening to identify high-expression signal peptides with minimal N-terminus biases via yeast display. ACS Synth Biol. 2022;11(7):2405–16.
Jarnot P, Ziemska-Legiecka J, Grynberg M, Gruca A. Insights from analyses of low complexity regions with canonical methods for protein sequence comparison. Brief Bioinform. 2022;23(5):bbac299.
Cosson P, Perrin J, Bonifacino JS. Anchors aweigh: protein localization and transport mediated by transmembrane domains. Trends Cell Biol. 2013;23(10):511–7.
Zhang Y, Zhang HZ, Fu JY, Du YY, Qu J, Song Y, et al. The GmXTH1 gene improves drought stress resistance of soybean seedlings. Mol Breed. 2021;42(1):3.
Nagashima Y, von Schaewen A, Koiwa H. Function of N-glycosylation in plants. Plant Sci. 2018;274:70–9.
Eklof JM, Brumer H. The XTH gene family: an update on enzyme structure, function, and phylogeny in xyloglucan remodeling. Plant Physiol. 2010;153(2):456–66.
Baumann MJ, Eklof JM, Michel G, Kallas AM, Teeri TT, Czjzek M, et al. Structural evidence for the evolution of xyloglucanase activity from xyloglucan endo-transglycosylases: biological implications for cell wall metabolism. Plant Cell. 2007;19(6):1947–63.
Sarmiento-Lopez LG, Lopez-Espinoza MY, Juarez-Verdayes MA, Lopez-Meyer M. Genome-wide characterization of the xyloglucan endotransglucosylase/hydrolase gene family in Solanum lycopersicum L. and gene expression analysis in response to arbuscular mycorrhizal symbiosis. PeerJ. 2023;11:e15257.
Rose JK, Braam J, Fry SC, Nishitani K. The XTH family of enzymes involved in xyloglucan endotransglucosylation and endohydrolysis: current perspectives and a new unifying nomenclature. Plant Cell Physiol. 2002;43(12):1421–35.
Sasidharan R, Chinnappa CC, Staal M, Elzenga JT, Yokoyama R, Nishitani K, et al. Light quality-mediated petiole elongation in Arabidopsis during shade avoidance involves cell wall modification by xyloglucan endotransglucosylase/hydrolases. Plant Physiol. 2010;154(2):978–90.
Matsui A, Yokoyama R, Seki M, Ito T, Shinozaki K, Takahashi T, et al. AtXTH27 plays an essential role in cell wall modification during the development of tracheary elements. Plant J. 2005;42(4):525–34.
Kushwah S, Banasiak A, Nishikubo N, Derba-Maceluch M, Majda M, Endo S, et al. Arabidopsis XTH4 and XTH9 contribute to wood cell expansion and secondary wall formation. Plant Physiol. 2020;182(4):1946–65.
Yang L, Machin F, Wang S, Saplaoura E, Kragler F. Heritable transgene-free genome editing in plants by grafting of wild-type shoots to transgenic donor rootstocks. Nat Biotechnol. 2023;41(7):958–67.
Loupit G, Brocard L, Ollat N, Cookson SJ. Grafting in plants: recent discoveries and new applications. J Exp Bot. 2023;74(8):2433–47.
Cookson SJ, Clemente Moreno MJ, Hevin C, Nyamba Mendome LZ, Delrot S, Magnin N, et al. Heterografting with nonself rootstocks induces genes involved in stress responses at the graft interface when compared with autografted controls. J Exp Bot. 2014;65(9):2473–81.
Ji P, Liang C, Yang Y, Wang R, Wang Y, Yuan M, et al. Comparisons of anatomical characteristics and transcriptomic differences between heterografts and homografts in Pyrus L. Plants (Basel). 2022;11(5):580.
Zhong R, Lee C, Ye ZH. Global analysis of direct targets of secondary wall NAC master switches in Arabidopsis. Mol Plant. 2010;3(6):1087–103.
He L, Xu J, Wang Y, Yang K. Transcription factor ANAC074 binds to NRS1, NRS2, or MybSt1 element in addition to the NACRS to regulate gene expression. Int J Mol Sci. 2018;19(10):3271.
Millard PS, Kragelund BB, Burow M. R2R3 MYB transcription factors - functions outside the DNA-Binding domain. Trends Plant Sci. 2019;24(10):934–46.
Noguero M, Atif RM, Ochatt S, Thompson RD. The role of the DNA-binding one zinc finger (DOF) transcription factor family in plants. Plant Sci. 2013;209:32–45.
Zhong R, Lee C, Ye ZH. Functional characterization of poplar wood-associated NAC domain transcription factors. Plant Physiol. 2010;152(2):1044–55.
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15(10):573–81.
Thomas H, Van den Broeck L, Spurney R, Sozzani R, Frank M. Gene regulatory networks for compatible versus incompatible grafts identify a role for SlWOX4 during junction formation. Plant Cell. 2022;34(1):535–56.
Zhong R, Richardson EA, Ye ZH. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. Plant Cell. 2007;19(9):2776–92.
Zou X, Sun H. DOF transcription factors: Specific regulators of plant biological processes. Front Plant Sci. 2023;14:1044918.
Zhang A, Matsuoka K, Kareem A, Robert M, Roszak P, Blob B, et al. Cell-wall damage activates DOF transcription factors to promote wound healing and tissue regeneration in Arabidopsis thaliana. Curr Biol. 2022;32(9):1883–94 e7.
Liu YB, Lu SM, Zhang JF, Liu S, Lu YT. A xyloglucan endotransglucosylase/hydrolase involves in growth of primary root and alters the deposition of cellulose in Arabidopsis. Planta. 2007;226(6):1547–60.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Letunic I, Khedkar S, Bork P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 2021;49(D1):D458–60.
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–52.
Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015;43(W1):W39–49.
Fu MM, Liu C, Wu F. Genome-wide identification, characterization and expression analysis of xyloglucan endotransglucosylase/Hydrolase genes family in barley (Hordeum vulgare). Molecules. 2019;24(10):1935.
Tamura K, Stecher G, Kumar S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38(7):3022–7.
Subramanian B, Gao S, Lercher MJ, Hu S, Chen WH. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019;47(W1):W270–5.
Roth C, Liberles DA. A systematic search for positive selection in higher plants (Embryophytes). BMC Plant Biol. 2006;6:12.
Nekrutenko A, Makova KD, Li WH. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res. 2002;12(1):198–202.
Zhao H, Jiang J, Li K, Liu G. Populus simonii × Populus nigra WRKY70 is involved in salt stress and leaf blight disease responses. Tree Physiol. 2017;37(6):827–44.
Hidvegi N, Gulyas A, Dobranszki J, Teixeira da Silva JA. Mining sequences with similarity to XTH genes in the Solanum tuberosum L. transcriptome: introductory step for identifying homologous XTH genes. Plant Signal Behav. 2020;15(10):1797294.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32101478, 32271830), the China Postdoctoral Science Foundation (2021M702360), and the Natural Science Foundation of Shandong Province (ZR2020QC033).
Author information
Authors and Affiliations
Contributions
L.Y. conducted the experiment, analyzed the data, and wrote the manuscript. Y.C. and X.L. collected some data. S.Z. gave some important suggestions and modifications for writing the manuscript. Q.H. (the corresponding author) had the overall responsibility for experimental design and project management. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
The characteristics of PtrXTHs and SpuXTHs. Supplementary Table 2. The Ka/Ks ratios of poplar and willow gene pair. Supplementary Table 3. The secondary structure prediction of PtrXTHs and SpuXTHs. Supplementary Table 4. The protein sequences of poplar and willow. Supplementary Table 5. Specific primers for XTH genes in poplar and willow. Supplementary Table 6. The predicted transcription factors that regulate five XTH genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, L., Chen, Y., Liu, X. et al. Genome-wide identification and expression analysis of xyloglucan endotransglucosylase/hydrolase genes family in Salicaceae during grafting. BMC Genomics 24, 676 (2023). https://doi.org/10.1186/s12864-023-09762-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09762-y