
Abstract
Background
Freezing temperatures are an abiotic stress that has a serious impact on plant growth and development in temperate regions and even threatens plant survival. The wild apple tree (Malus sieversii) needs to undergo a cold acclimation process to enhance its freezing tolerance in winter. Changes that occur at the molecular level in response to low temperatures are poorly understood in wild apple trees.
Results
Phytohormone and physiology profiles and transcriptome analysis were used to elaborate on the dynamic response mechanism. We determined that JA, IAA, and ABA accumulated in the cold acclimation stage and decreased during freezing stress in response to freezing stress. To elucidate the molecular mechanisms of freezing stress after cold acclimation, we employed single molecular real-time (SMRT) and RNA-seq technologies to study genome-wide expression profiles in wild apple. Using the PacBio and Illumina platform, we obtained 20.79G subreads. These reads were assembled into 61,908 transcripts, and 24,716 differentially expressed transcripts were obtained. Among them, 4410 transcripts were differentially expressed during the whole process of freezing stress, and these were examined for enrichment via GO and KEGG analyses. Pathway analysis indicated that “plant hormone signal transduction”, “starch and sucrose metabolism”, “peroxisome” and “photosynthesis” might play a vital role in wild apple responses to freezing stress. Furthermore, the transcription factors DREB1/CBF, MYC2, WRKY70, WRKY71, MYB4 and MYB88 were strongly induced during the whole stress period.
Conclusions
Our study presents a global survey of the transcriptome profiles of wild apple trees in dynamic response to freezing stress after two days cold acclimation and provides insights into the molecular mechanisms of freezing adaptation of wild apple plants for the first time. The study also provides valuable information for further research on the antifreezing reaction mechanism and genetic improvement of M. sieversii after cold acclimation.
Similar content being viewed by others


Background
Central Asian mountains are particularly important and have been listed as a global biodiversity hotspot. These hotspots contain ancestors of some domestic fruit, including apples. Malus sieversii (also called “**njiang wild apple”), the ancestor of domesticated apple [1], is mainly distributed in the Tianshan Mountains of China, Kazakhstan, Kyrgyzstan, and Uzbekistan. M. sieversii is a germplasm resource with excellent cold resistance traits and is usually used as a popular rootstock for breeding cold-resistant domesticated apple in China [2, 3]. Therefore, it is of great significance to understand the cold resistance mechanism of wild apple for its further protection and utilization.
As an abiotic stress, low temperature not only affects the geographical distribution of crops, but also has a negative impact on crop yield and quality. Cold stresses are divided into chilling (0–15 °C) and freezing (< 0 °C) damage, can lead to tissue damage and even plant death, and often bring large economic losses to agricultural production [4]. When plants are exposed to low temperatures, the cell membrane is the main site of cold-induced damage, which triggers a series of physiological and biochemical reactions [5]. After exposure to low temperature for a period of time, many plants can improve frost resistance, a process called cold acclimation (CA) [6]. During cold acclimation, plants are able to sense environmental temperature changes, and then trigger a series of protective mechanisms, including changing the composition, structure and function of membrane; synthesis of cryoprotectant molecules, such as soluble sugar, protein and proline; increase the activity of protective enzymes [7]. CA can also induce the expression of some cold-induced genes during the stress period, and these genes likely play a role in stabilizing the cell membrane against freezing injury [8]. For example, CBF/DREB1 protein from Arabidopsis transcription factor family has been identified to control the expression of a regulon of cold-induced genes that increase plant freezing tolerance [9]. In apple (Malus domestica), homologue MdCIbHLH1 of Arabidopsis ICE1 contributes to improved cold tolerance by directly activating CBF genes [10]. Furthermore, it is known that the MYB-bHLH-WDR complex, especially the bHLH and MYB components, is involved in modulating anthocyanin accumulation at low temperatures [11]. Additionally, the accumulation of sucrose and other monosaccharides during cold acclimation also contributes to membrane stability because these molecules can protect the membrane from freezing damage in vitro [12]. The entire CA process in nature typically requires much time and is easily affected by climate fluctuations. However, with global warming, the winters and early springs in temperate regions are becoming progressively milder, and temperature patterns are becoming increasingly irregular. This increases the frequency of temperature changes that may cause premature subzero temperatures, thereby subsequent increasing the risk of freezing injury [13]. Additionally, changing phenological patterns, such as an early start of growing season and early flowering time [14], consistent with climate warming, it may increase the risk of tissue damage caused by frost. The likelihood of such scenarios is typically high during early spring. Therefore, it is very important to understand the mechanism of freezing stress after cold acclimation to protect and utilize plants. However, little is known about the molecular mechanism of freezing stress after a short period of cold acclimation in M. sieversii.
According to previous research, such as on Hordeum vulgare [15], Arabidopsis thaliana [16], Glycine max [17], and Camellia sinensis [18], the molecular mechanism of plants responding to freezing stress has been explained. Transcriptome sequencing was used to obtain differentially expressed genes (DEGs) and further explore the role of related signaling pathways and candidate genes to explain the changes in the plant response to freezing stress. For instance, strawberry (Fragaria×ananassa) responds to freezing stress through several pathways: flavonoid biosynthesis, plant hormone signal transduction, MAPK (mitogen-activated protein kinase) signaling, starch and sucrose metabolism, circadian rhythm [19]. A previous transcriptome study identified 4173 and 7734 DEGs in two apple cultivars, respectively, during the chilling and freezing treatments [20]. Oil rapeseed (Brassica napus) responds to low-temperature stress through two conserved (primary metabolism and plant hormone signal transduction pathway) and two novel (plant-pathogen interaction and circadian rhythm pathway) signaling pathways [21]. Transcriptomic analysis of Magnolia wufengensis under cold stress showed that the response mechanism was related to photosynthesis, and plant hormone signal transduction, primary and secondary metabolism pathways [22].
RNA-seq based on the Illumina platform is a powerful method for investigating gene expression profiles and revealing signal transduction pathways in a wide range of biological systems [23]. In recent years, RNA-seq technology has been widely used to clarify the response of various plants to low temperature stress, including C. sinensis [24], and A. thaliana [25]. In apple (Malus domestica), comparative transcriptome analysis revealed 377 commonly upregulated and 211 commonly downregulated DEGs involved in the crosstalk between drought, cold and high-salinity stress [26]. Single molecule real-time (SMRT) sequencing based on PacBio (Pacific Biosciences) platform can capture the full length of transcripts, which provides a simpler and more accurate method for gene annotation and isoform identification. The third generation sequencing technology represented by PacBio has the advantage of long read length [27]. PacBio and RNA-seq are highly complementary to each other. To obtain an overall view of the molecular regulation during freezing stress after the CA process, we combined PacBio sequencing and RNA-seq to investigate the transcriptome of M. sieversii seedlings at low temperatures. This study will provide a theoretical basis for freezing stress resistance and lay a foundation for understanding the cold response molecular mechanism in M. sieversii, which provides a rich genetic resource for further research investigating the freezing response in apple and other woody plants.
Results
Physiological and plant hormone changes of M. sieversii under low temperature stress
In this study, 8-week-old apple plantlets were used as experimental materials to study the transcriptomic and physiological changes of M. sieversii under freezing stress (Fig. 1A). The well-developed wild apple seedlings were cold acclimated at 4 °C in a chamber for two days. Subsequently, the temperature was gradually lowered from 4 °C to − 4 °C (2 °C/h) for 4 h, and samples were collected at 0, 1, 6, and 10 h intervals at − 4 °C (Fig. 1B). To evaluate the physiological damage caused by low temperature, we measured relative electrolyte leakage (REC) values, maximum photosynthetic efficiency (Fv/Fm) values, malondialdehyde (MDA) content, and sucrose content. The plants treated at − 4 °C showed a relatively higher sucrose content, which was approximately 200% higher during the 1 h interval (Fig. 1C). Furthermore, under low-temperature treatment, the Fv/Fm values were significantly reduced compared with those of the control (Fig. 1E). In addition, the REC values and MDA production in the treated group were relatively higher than those in the control (Fig. 1D, F), indicating that plasma membranes may be damaged under freezing stress. Low-temperature treatment significantly affected the endogenous hormone content of **njiang wild apple leaves. Compared with the control, the contents of endogenous jasmonic acid (JA) and abscisic acid (ABA) after cold acclimation (0 h) increased significantly. Compared with cold acclimation (0 h), the contents of endogenous JA and ABA in the treated leaves showed a downward trend (0–10 h), but these values were still higher than those of the control (Fig. 1G,H), except for the ABA content at 10 h. Moreover, we also determined the content of IAA (Fig. 1I). Surprisingly, the content of IAA in the cold acclimation stage (0 h) did not decrease, but under frozen stress (0 h–10 h), the content decreased significantly. In general, cold and freezing environments produce different hormonal responses.

Processing methods and physiological and biochemical indicators after cold treatments at various time points. (A) Two-month-old wild apple seedlings growing in tissue culture bottles (left); harvested leaf (right); (B) Treatment process and sampling time points; (C) sucrose content; (D) MDA content; (E) Fv/Fm values; (F) REC values; (G) endogenous ABA content; (H) endogenous JA content; and (I) endogenous IAA content. Bars represent the mean ± SD (n = 3). Accumulation levels were analyzed using a T-test. Within each figure, asterisks above the bars indicate statistical significance (*p < 0.05; **p < 0.01)
Analysis of sample Iso-Seq results
To identify and characterize the transcript dynamics of M. sieversii under freezing treatment after cold acclimation, we employed PacBio SMRT and RNA-seq technologies for transcriptome sequencing. In this study, the sample materials use for transcriptomic analysis and the treatment methods were consistent with the physiological measurements in Fig. 1. Subsequently, the cDNA libraries constructed from treated and CK samples were sequenced. After correcting the data, a total of 11,781,904 (20.79G) subreads were obtained (Table 1). In addition, a total of 611,407 circular consensus sequence (CCS) reads were obtained after filtering. Finally, 485,006 full-length non-chimeric reads (FLNC) were obtained and poly-A tails in which an average FLNC read length was 1765 bp. In total, 432,508,752 nucleotides were obtained, and 191,961 reads were generated. The minimum length was 193 bp, and the maximum length was 14,168 bp. The N50 value was 2613 bp, and the N90 value was 1344 bp. The length of each subread sequence was counted after quality control of the raw data, and the length distribution map was generated (Additional file 1).
GMAP software was used to match the consensus sequences to the reference genome sequences, and a total of 176,234 (91.81%) reads were mapped to the Malus domestica genome sequences (GDDH13 v1.0); according to the results, the sequences can be divided into four types: unmapped, 15,727 (8.19%); multiple mapped, 10,438 (5.44%); reads mapped to the forward strand (+), 95,421 (49.71%); and reads mapped to the antisense strand (−), 70,375 (36.66%) (Fig. 2A). To obtain a comprehensive genetic annotation, the functions of novel genes were annotated based on seven databases. Of the 3917 novel genes, 3808 were significantly matched in at least one of the databases. Among them, 3359, 2242, 3216, 1625, 2038, 3708, and 2038 novel genes were found in the NR (NCBI nonredundant protein sequence), SwissProt, KEGG (Kyoto Encyclopedia of Genes and Genomes), KOG/COG (Clusters of Orthologous Groups of proteins), GO (Gene Ontology), NT (NCBI nucleotide sequences by BLAST, E-value 1e-10), and Pfam (Protein family) databases, respectively (Additional file 1). The histogram demonstrated that 1097 genes were simultaneously annotated across all seven databases. We compared the 61,908 transcripts with the M. domestica genome gene set, and they were classified into three groups, with following results are shown in Fig. 2B: isoforms of known genes (13,069, 21.11%), novel isoforms of known genes (43,794, 70.74%), and isoforms of novel genes (5045, 8.15%). Alternative splicing (AS) events were analyzed with SUPPA software. We detected 22,316 transcripts with alternative splicing (AS) events, including skipped exons (SE), mutually exclusive exons (MX), alternative 5′splice site (A5), alternative 3′splice site (A3), retained intron (RI), alternative first exon (AF) and alternative last exon (AL) (Fig. 2C, Additional file 6). The transcripts were analyzed by principal component analysis (PCA) (Additional file 2). The first two axes of the PCA represent the differentiation between the biological replicates of the different treatments. In addition, 1710 fusion transcripts were identified (Additional files 3, 7).

Characterization of M. sieversii transcripts. (A) GMAP map** statistics. (B) Classification of transcripts corrected based on the reference genome sequences. (C) The gene numbers involving AS events. TML represents mixed samples containing treated and control samples
Identification and analysis of DETs
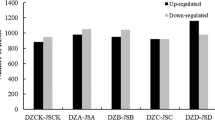
In total, we obtained 24,716 DETs in different comparisons (Additional file 8). To better understand the gene expression changes between the CK and cold-treated apple leaf samples in response to freezing after 2 d of cold acclimation, we compared the number of DETs that were different among time points (0 h vs CK, 1 h vs CK, 6 h vs CK, 10 h vs CK, 10 h vs 0 h). A total of 6430 DETs were obtained between 0 h and CK, with 2493 upregulated and 3487 downregulated transcripts; additionally, 7463 (3590 upregulated and 3873 downregulated), 7133 (3711 upregulated and 3422 downregulated), 7192 (3868 upregulated and 3324 downregulated), and 570 (405 upregulated and 165 downregulated) transcripts were identified among the 1 h vs CK, 6 h vs CK, 10 h vs CK, 10 h vs 0 h comparisons, respectively (Fig. 3A). We used the Venn diagram to identify DETs that responded continuously during freezing stress. From the Venn results, a total of 2234 DEGs and 4410 DETs (2457 upregulated and 1953 downregulated) were shared across all treatments (Fig. 3B, Additional files 9, 10). We conducted a GO enrichment and KEGG pathway analysis on all upregulated and downregulated DETs to explore their biological functions (Additional files 11, 12, 13). In addition, KEGG pathway analyses were also conducted to identify several important pathways (Fig. 3C-E, Additional file 4,). In the pairwise, DETs were significantly enriched in the “biosynthesis of secondary metabolites”, “photosynthesis”, “peroxisome”, “starch and sucrose metabolites” and “plant hormone signal transduction” categories. All the pathways worked together to form a complex and efficient signaling network that reduced or even eliminated cold stress damage. The pathway enrichment results indicated that the major pathways included plant hormone signal transduction, starch and sucrose metabolism, peroxisomes, transcription factors, and photosynthesis.

Analysis of differentially expressed transcripts (DETs) at different times (CK, 0, 1, 6, and 10 h) across the transcriptome. (A) Number of DETs in the pairwise analyses. Upregulated (orange) and downregulated (blue) transcripts were quantified. (B) Venn diagram of the DETs at the different time points compared with the CK; (C) KEGG pathway enrichment at 0 h vs CK; (D) KEGG pathway enrichment at 10 h vs CK; (E) KEGG pathway enrichment at 10 h vs 0 h. dpi. A rich factor was the ratio of an input number to the background number in a specific pathway. The size and color of the dots represent the transcript numbers and the q-values, respectively
Sugar and starch metabolism pathway
The sugar and starch metabolism pathway (Fig. 4A) showed that two related starch synthase 2 (SS2) genes (MD09G1052000 and MD00G1130900) were downregulated. However, another SS1 gene (MD01G12218000) was upregulated. Sucrose synthase 3 (SUS3) gene (MD11G1307000, MD11G1307000_novel20) was upregulated. Notably, the gene expression of the Bate-amylase (BAM) family members (MD16G1231500, MD16G1231500_novel09, MD06G1044200, MD06G1043900, Novelgene2490, Novelgene2490_novel01, MD07G1287700, and MD06G1044100) was significantly upregulated under freezing stress. It is worth noting that the UDP glucose 6-dehydrogenase genes (UGDH) (MD16G1062900, MD13G1064400, MD13G1064400_novel01), which catalyze UDP-alpha-D-glucuronate, exhibited inducible expression. In short, there are up and downregulated genes across various biological reactions that may occur in wild apple plants in response to cold stress.

Heat map of the relative expression levels of DEGs involved in sugar metabolism and the antioxidant defense system; (A) Sugar and starch metabolism-related genes; (B) Antioxidant-related genes. The red and blue colors indicate high and low expression levels (log2 fragments per kilobase of transcript per million mapped reads, FPKM), respectively
Antioxidant defense system
According to the heat map of the antioxidant-related gene expression levels of DEGs (Fig. 4B), superoxide dismutase 1 (SOD1) (MD00G1051500, MD00G1051500_novel02), L-ascorbate peroxidase T (APXT) (MD00G1029300), peroxidase (POD) (MD05G1075100, MD03G1012700, MD10G1206100, MD04G1171700, MD03G1059200, MD01G1162100), thioredoxin (TRX) (MD05G1082500, MD13G1080800), respiratory burst oxidase homolog D (RBOHD) (MD13G1134500, MD13G1134500_novel02, MD13G1134500_novel10), and nucleoredoxin (NRX) (MD13G1093200, MD16G1094400, MD16G1094400) were downregulated. However, glutathione S-transferase U7 (GSTU7) (MD10G1172100), PER (MD17G1040100, MD13G1134500, and MD13G1086200), glutathione transferase 23 (GST23) (MD10G1172300), and catalase isozyme1 (CAT1) (MD06G1008700) genes were significantly upregulated under freezing stress conditions.
Responses of the plant hormone signaling network to cold stress
In the ABA signal transduction pathway (Fig. 5A), 2 PYL genes (MD08G1043500, MD12G1178800, MD12G1178800_novel01) were upregulated under freezing stress conditions, whereas another PYL gene (MD05G1300200), a protein phosphatase 2C (PP2C) gene (MD01G1139200, MD01G1139200_novel04), and two ABF genes (MD15G1081800, MD15G1081800_novel02, MD15G1081800_novel03, MD08G1099600, MD08G1099600_novel01) were downregulated. In the JA signal transduction pathway (Fig. 5B), several TIFY proteins (MD02G1096100, MD06G1228900, MD09G1178600, MD10G1260700, MD15G1116100, MD15G1116100_novel01, MD16G112740, and MD17G1164400) were induced under freezing stress in this study. In the BR signal transduction pathway (Fig. 5C), BRASSINAZOLE RESISTANT1 (BZR1) (MD15G1031900, MD08G1035400), BRI1-associated kinase 1 (BAK1) (MD04G1054400), and Cyclin D3 (CYCD3) (MD15G1288100) were downregulated. In the phytohormone ethylene (ET) signal transduction pathway (Fig. 5D), serine/threonine-protein kinase 1 (CTR1) (MD09G1236700, MD10G1168800) and ETHYLENE INSENSITIVE 3 (EIN3) (MD02G1266200), and ethylene-responsive transcription factor (ERF1) (MD04G1228800, MD04G1228800_novel01) were significantly upregulated under freezing stress. In the auxin signal transduction pathway (Fig. 5E), TRANSPORT INHIBITOR RESPONSE (TIR) (Novelgene0397), IAA/indole/− 3-acetic acid (AUX/IAA) (MD10G1176400, MD13G1204700, MD17G1183500, MD17G1198300, MD09G1216100), and small-IAA-up-RNA (SAUR) (MD07G1297400, MD07G1297400_novel01, MD16G1240600) were upregulated, whereas ARF (MD01G1083400, MD14G1131900) and SAUR (MD10G1059800, MD05G1113200, MD05G1113200_novel01, MD00G1067200, MD15G1014200, MD10G1061300, MD16G1240600) were downregulated.

Plant signal transduction pathways and expression of related genes in M. sieversii. (A) Heatmap of the ABA signal transduction pathway. (B) Heatmap of the JA signal transduction pathway. (C) Heatmap of the BR signal transduction pathway. (D) Heatmap of the ET signal transduction pathway. (E) Heatmap of the auxin signal transduction pathway. The red and blue colors indicate high and low expression levels (log2 fragments per kilobase of transcript per million mapped reads, FPKM), respectively
Photosynthesis
The KEGG enrichment analyses of DETs showed that the photosynthesis pathway was significantly enriched. We focused our attention on photosynthesis-related DETs and found that most of these transcripts were downregulated under cold conditions. In PSI, the DEGs photosystem I light reaction center (MD01G1004300, MD17G1191200, MD17G1191400, MD13G1191300, MD03G1042800, MD10G1238800, MD16G1191400), photosystem I subunit O (PsaO) (MD03G1150500, MD03G1150500_novel01), and ATP synthase subunits (ATPase) (MD05G1347300, MD17G1003000) were downregulated. In PSII, the DEGs oxygen-evolving enhancer protein 2 (OEEP2) (MD17G1132900, MD07G1 190900, MD07G1191200, MD09G1146300), oxygen-evolving enhancer protein 3 (OEEP3) (MD10G1311700, MD11G1227900), PSII light reaction center (MD10G1176000), PsbW gene (MD10G1192100), and PsbY genes (MD16G1106800, MD16G1106800_novel01, MD13G1106700, MD13G1106700_novel01) were downregulated. However, two of the DEGs related to photosynthesis were upregulated, PSII core complex gene psbY (MD06G1160500) and PSII reaction center gene PSB28 (MD10G1176000), during low-temperature stress in M. sieversii. The results are shown in Table 2.
Transcription factors (TF)
TFs are known to play essential roles in plant responses to abiotic stress. The iTAK and HMMER analysis of apple transcripts allowed the identification of nonredundant TFs. In our work, we analyzed the expression patterns of 1801 transcripts of 676 genes in the top 15 transcription factor families (Fig. 6, Additional file 14). Heatmaps of abundance-altered TFs were plotted to depict the transcript abundance profiles of each TF family (Fig. 6B, C). The MYB superfamily was the largest, including 154 MYB and 178 MYB-related members. The second-largest family was the bHLH family, with 161 members, followed by the bZIP (143), C2H2 (136), C3H (136), WRKY (118), SNF2 (112), GARP-G2-like (112), PHD (102), NAC (98), AP2-ERF (92), SET (108), HB-HD-ZIP (82), and Jumonji (66) families.

Identification and analysis of the transcript expression levels of the top 15 transcription factor families. (A) Statistics of the differentially expressed transcription factors. (B) Expression profiles of differentially expressed TFs among different samples. The heat map was generated from the FPKM values. The red and blue colors indicate high and low expression levels, respectively
Validation of DETs by qRT-PCR analysis
Sixteen DETs related to cold adaptation in different signaling pathways and TFs were randomly selected to validate the gene expression levels using qRT-PCR. They represented various functional categories or pathways, including sugar metabolism (SS1, SUS3), antioxidant defense systems (CAT1, GPX6), plant hormone signal transduction (PP2C, ABF2, ARF9, IAA17), TFs (NAC104, ERF3), and photosynthesis systems (psaD, PSB27, Cyt b6-f). Nine DETs showed elevated expression levels, and the expression levels of seven DETs decreased. The results show that the expression patterns were clearly consistent between qRT-PCR and transcriptome sequencing (Additional file 5). The independent qRT-PCR results confirmed the accuracy and reliability of the RNA-seq results.
Discussion
Apple is one of the most important fruit crops worldwide. M. sieversii, a progenitor of domesticated apple, represents a reservoir of genetic diversity and is widely used as a rootstock in China [28]. The selection of rootstocks is usually based on stress tolerance [29]. Cold stress is one of the most severe abiotic stresses in apple plants and is one of the many limiting factors for global apple production. Through cold acclimation, plants can alleviate the harm of low temperatures and enhance their tolerance to cold stress [30]. In tea plants, differential changes in transcription and metabolism levels occurred in response to CA and cold stress conditions [31]. Since the cold-stress response network is very complex and contains an array of biochemical and physiological modifications, many defense mechanisms are activated during cold acclimation, thus the transcriptome changes will be less intense even when the plant is exposed to lower temperatures. To discover the key functional and regulatory genes of cold resistance in M. sieversii, we examined seedlings under cold stress after 2 d of cold acclimation and performed full-sequence transcriptomic analysis. Full-sequence transcriptomic data have great potential for discovering new or previously unrecognized isoforms, meanwhile, different expression patterns appearing between isoforms of the same gene. For example, in present work, isoforms MD05G1126500_novel01, MD08G1162500_novel01, MD12G1040700_novel07, MD12G1126100_novel02, MD05G1126500_novel01, MD06G1228000 are up-regulated, another isoforms of the corresponding gene MD05G1126500_novel02, MD08G1162500, MD12G1040700_novel06, MD12G1126100_novel08, MD05G1126500_novel02, MD06G1228800_novel01 are down-regulated. In addition, the transcriptome results revealed that isoforms involved in transcription regulation, signal transduction of hormones, transcription factors, and oxidative stress were differentially expressed under cold stress, we also compared the activities of MDA and Fv/Fm and the contents of ABA and JA in seedlings of M. sieversii. To further explore the response mechanism of apple under freezing stress, we identified a series of genes from different pathways. This study provides a novel understanding of the roles of CA in freezing stress in M. sieversii.
Sugar and starch metabolism
The metabolism of sugar and starch plays a vital role in the cold resistance of plants. Sugar can be used as a signaling molecule in hormone synthesis, plant growth and development, and various antistress reactions, as a nutrient to supply energy and as a protective agent to avoid cold damage [32, 33]. SUS is a key enzyme involved in sucrose metabolism and is part of a family of genes encoding different sucrose isozymes, including SUS1 and SUS3 (MD11G1307000, MD02G1100500) were upregulated. Here, the induction of two genes encoding SUS may point towards the degradation of sucrose to UDP-glucose and fructose. UGDH (MD16G1062900, MD13G1064400) was induced. Under low-temperature stress, the task of increasing glycolysis can be completed [34]. Starch hydrolysis was associated with increased maltose accumulation and increased expression of BAM, which encodes the beta-amylase necessary for starch mobilization and raises the levels of soluble sugars capable of acting as osmolytes or antioxidants [35,36,37]. Under cold stress conditions, the activity of BAM1 was largely unaffected, but BAM3 expression increased [38]. In this study, two related SS genes (MD09G1052000 and MD00G1130900) were repressed, and another SS1 (MD01G1218000) was induced. Notably, the gene expression of BAM family members, such as MD16G1231500, MD06G1044200, MD06G1043900, and MD06G1044100, was significantly upregulated under freezing stress (Fig. 4A). This phenomenon may indicate that **njiang wild apples respond to freezing stress by accelerating the decomposition of starch to provide energy and achieve heat preservation.
Antioxidant defense system
In the antioxidant defense system, various nonenzymatic and enzymatic ROS detoxification mechanisms have developed in plant cells. Major enzymatic systems include superoxide dismutase (SOD), ascorbate peroxidase (APX), peroxidase (POD), and catalase (CAT) [39]. MDA levels and electrolyte permeability are commonly used as indicators of membrane structural damage and plant resilience [40, 41]. Under abiotic stress, antioxidant enzymes can eliminate harmful substances to the greatest extent and effectively reduce membrane damage. Plants with suppressed APX production induce SOD and CAT to compensate for the loss of APX, whereas plants with suppressed CAT production induce APX and GPX. In addition, POD and CAT are hydrogen peroxide scavengers and catalyze the conversion of hydrogen peroxide (H2O2) into water. In this study, genes encoding CAT (MD06G1008700), GST (MD10G1172300), GPX6 (MD06G1081300), APXT (MD04G1104900), SPS1 (MD10G1258500) and RBOHD (MD13G1134500, MD13G1134500_novel02, MD13G1134500_novel10) were upregulated under freezing stress conditions. However, the genes encoding POD (MD03G1013200, MD10G1206100, MD04G1171700, MD03G1059200, and MD01G1162100) were significantly downregulated (Fig. 4B). Overall, the antioxidant defense system-related genes also rapidly responded to freezing stress in **njiang wild apples, enabling plants to cope with freezing stress. Antioxidant defense systems involve many kinds of antioxidant genes, which have unique functions under stress conditions, forming a complex antioxidant defense system in vivo.
Responses of the plant hormone signaling network to cold stress
We measured ABA accumulation at − 4 °C for different times after cold acclimation. When cold acclimation ended, a large amount of ABA accumulated. ABA levels were increased in plants that were stressed, such as those under drought, cold, or high temperature stresses, and ABA played a crucial role in plant stress resistance [42,43,44]. During cold acclimation, ABA accumulation can induce the expression of some cold-resistant genes to improve freezing resistance [45]. Numerous studies found a direct link between the accumulation of ABA in woody plants and the increase in cold tolerance [46]. Important components of ABA include PP2C and PYL, which is closely related to the level of plant stress resistance [47, 48]. PYL (MD05G1300200) and PP2C (MD01G1139200, MD01G1139200_novel04) were induced in response to cold stress. ABF2 regulates various aspects of the ABA response by controlling the expression of a subset of ABA-responsive genes [49, 50]. In this study, ABF (MD08G1099600, MD08G1099600_novel01, MD15G1081800, MD15G1081800_novel02, MD15G1081800_novel03) was upregulated under freezing stress conditions. These results indicated that ABA stimulated this signaling pathway and that DEGs were involved in the ABA signaling response to freezing stress.
We also measured the content of JA, which show a consistent trend as that for ABA content. After cold acclimation (0 h), the content of JA increased significantly, while the content of JA decreased during the freezing treatment (0 h–10 h). However, the JA content was still higher than that of the control. Similar to ABA, cold stress induces a number of JA biosynthetic and signaling genes that ultimately trigger the accumulation of JA and increase cold tolerance by regulating the ICE-CBF/DREB1 cascade [51]. Consistent with the positive role of JA in freezing stress, the production of endogenous JA was triggered by cold treatment. Exogenous application of MeJA significantly improved plant freezing tolerance with or without cold acclimation [52]. In JA signal transduction pathway, JAZ genes (MD02G1096100, MD06G1228900, MD09G1178600, MD10G1260700, MD15G1116100, MD15G1116100_novel01, MD16G112740, and MD17G1164400) was upregulated under freezing stress in this study. The bHLH transcription factor MYC2 (MD01G1086900, MD06G1119900, and MD11G1254100) acted as a direct target of the JAZ protein to prevent JA signaling. In this study, JAZ protein was induced under freezing stress, whereas the transcription factor MYC2 was repressed. Taken together, these results indicated that the JA signaling pathway regulated **njiang wild apple freezing tolerance after cold acclimation.
BR and ET also play major roles in plant defense responses to cold stress [53]. It is worth noting that in BR signal transduction pathways, BAK1 (MD04G1054400), a transmembrane receptor kinase; BZR1 (MD15G1031900, MD08G1035400), a core member of the BR signaling pathway; and CYCD3 (MD15G1288100) were repressed under cold stress (Fig. 5). ET levels increase in response to cold stress and can induce the accumulation of antifreeze proteins [54]. In this study, genes encoding CTR1 (MD09G1236700, MD10G1168800), EIN3 (MD02G1266200), and ERF1 (MD04G1228800, MD04G1228800_novel01) were upregulated. In the ET signaling pathway, CTR1 is a positive regulator in the ethylene signaling pathway. EIN3 is controlled by a ubiquitin proteasome system, and ERF1 is an immediate target for EIN3 [55]. The results indicated that these genes in the ethylene signaling transduction pathway responded to freezing stress. These results indicate that both the BR and ET signaling pathways may function in M. sieversii cold response processes after acclimation.
In particular, the IAA content did not decrease significantly during cold acclimation but decreased significantly during freezing stress. We demonstrated that the endogenous hormone IAA may have a special role in cold acclimation. Moreover, the decreasing trend of the IAA content under freezing stress is similar to that of ABA and JA, which may involve coordination with ABA and JA. Although the role of auxin after cold stress is not clear, genes related to the IAA signal transduction pathway were differentially expressed in this study. IAA (MD10G1176400, MD13G1204700) and SAUR (MD12G1027600, MD07G1297400, MD07G1297400_novel01) were induced in response to low temperature (Fig. 5E), whereas ARF (MD01G1083400, MD14G1131900) and SAUR (MD10G1059800, MD10G1061300) were repressed. Analysis of genes related to the IAA signal transduction pathway showed that the TIR1 (Novelgene0397) transcript was highly expressed. TIR1 can cause AUX/IAA ubiquitination, relieving the inhibition of ARF activity by AUX/IAA and hence initiating the transcription of IAA-induced genes. Recent studies have shown that plant changes induced by cold stress are closely related to the auxin response [56,57,58]. These genes were differentially expressed in response to low temperature, indicating that the auxin signaling pathway may participate in the M. sieversii cold response. In conclusion, the M. sieversii freezing response after CA involves a complicated regulatory network containing multiple types of plant hormone signaling pathways.
Photosynthesis
The direct impact of abiotic stress on the activity of photosynthesis was the disruption of all photosynthetic components, such as photosystems I and II, electron transport, carbon fixation, ATP generation, and stomatal conductance [59], which resulted in serious damage to the photosynthetic machinery of plants [60]. In this study, DEGs associated with photosynthesis showed that most of these genes were downregulated, such as PsaN (MD01G1004300), PsaL (MD04G1244400, Novelgene0781), PsaO (Novelgene3166), PsbP (MD17G1132900, MD09G1146300), PetC (MD09G1098300, MD09G1098300_novel02), and ATPc (MD05G1347300). These results suggest that photosynthesis may be inhibited by freezing conditions. It is worth noting that two genes were upregulated, PSII core complex gene psbY (MD06G1160500) and PSII reaction center gene PSB28 (MD10G1176000). The evidence of genes that encode proteins linked to photosynthesis in the previous study as psbY is indicating that the slowdown in the degradation of chlorophylls by 1-MCP would also affect other photosystem II-related proteins [61]. PsbY gene up-regulation expression may play a critical role in photosynthetic systems under freezing stress. Low temperatures also cause a steady-state light imbalance, which reduces the quantum efficiency of PSII. This implies that freezing stress could induce a series of changes in genes related to photosynthetic pathways, thus decreasing Fv/Fm values.
Transcription factor dynamics during freezing responses
In this study, 1801 transcripts of 676 TFs from the top 15 TF families responding to freezing stress were examined (Fig. 6A), and the expression patterns of the top 15 TF families were analyzed (Fig. 6B,C). Many of these families of TFs were reported to play central roles in plant responses to abiotic stress, including cold stress [62,63,64,65,66]. A total of 49 AP2/ERF members were detected under freezing stress. DREB1/CBF (MD04G1067800, MD04G1067800_novel01, MD04G1067800_novel02, MD04G1165400, MD04G1165400_novel01), a subfamily member of AP2/ERF, was identified to be induced at least tenfold and twofold under freezing stress, respectively. Similarly, in previous studies, the expression of the CBF gene was upregulated in apple bark at different freezing temperatures [20]. Similarly, MYB113 (MD09G126510), MYB1 (MD09G1278600, MD09G1278600_novel02), MYB62 (MD13G1039900), MYB108 (MD13G1148200, MD13G1148200_novel01) and MYB4 (MD16G1218900) strongly responded to freezing stress after cold acclimation, and this response continued throughout the whole freezing stress period. In previous studies, three MYB transcription factors (MdMYB12, MdMYB22, and MdMYB114), which had several CBF/DREB response elements in their promoters, were significantly induced by low-temperature exposure and their expression also correlated highly with anthocyanin accumulation [11]. In apple, MYB transcription factors, such as MYB1 and MYB113, MYB114 are involved in the regulation of anthocyanin synthesis pathways [67, 68]. During freezing stress, low temperature induces the expression of MYB transcription factors and enhances anthocyanin synthesis to enhance freezing resistance. In addition, recent investigations in apple show that MdMYB4-transgenic apple calli exhibited enhanced tolerance to cold stress, as indicated by reduced electrolyte leakage value, and MDA content than the control calli [69]. The upregulation of MYB4 may increase cold resistance in wild apples. It also appears likely that WRKY TFs play important roles in M. sieversii cold regulation. Under freezing stress, 118 WRKY members were differentially expressed, among which WRKY71 (MD10G1266400, MD10G1266400_novel01), WRKY6 (MD10G1324500), and WRKY70 (MD12G1189900) were induced at least 5-fold. In Arabidopsis, WRKY70 in promoting BR-mediated gene expression to regulated plant growth and drought response, WRKY71 activity hastens flowering, thereby providing a means for the plant to complete its life cycle in the presence of salt stress [70, 71]. In wild apple, the high expression level of WRKY70 and WRKY71 may affect plant growth to cope with low temperature environment. Our transcriptome data also showed that other transcription factors, such as bHLH (161), bZIP (143), C2H2 (136), C3H (136), WRKY (118), SNF2 (112), GARP-G2-like (112), PHD (102), NAC (98), AP2-ERF (92), SET (108), HB-HD-ZIP (82), and Jumonji (66) families, were induced. These important abiotic stress response regulators may function under cold stress conditions in M. sieversii, providing important genetic resources for cold-tolerant domesticated apple breeding.
Conclusion
In this study, we first present the transcriptome of M. sieversii using PacBio single-molecule long-read sequencing to analysis of the responses to freezing temperature after cold acclimation. The expression patterns of 4410 DETs were determined, and identify several important pathways genes such as genes encoding superoxide dismutase, sucrose synthase, plant hormone-signaling transduction kinase, and peroxidase. In addition, a total of 1801 transcripts were confirmed as TFs. Some different members of the AP2/ERF, WRKY, MYB, and bHLH gene families were significantly up/downregulated. Furthermore, the transcription factors DREB1/CBF, MYC2, WRKY71, WRKY6, WRKY70, MYB4 and MYB88, were strongly induced during the whole stress period. In short, cold acclimation can induce the continuous expression of some antifreeze genes during freezing stress. These findings provide a characterization of gene transcription and facilitate the understanding of the mechanisms of tolerance to low temperature after cold acclimation in M. sieversii.
Methods
Plant materials
Seeds of M. sieversii were collected in September 2016 from one area (43°23′2.20″N; 83°35′43.48″E) in a natural wild reserve forest in Yili, **njiang. With the permission of the **nyuan County Forestry Bureau, seeds were obtained from the field and identified [72]. The field investigation and sample collection experiments were performed under institutional guidelines in accordance with local legislation. The germplasm of M. sieversii was identified by Ph.D. Wenjun Li, who worked at the **njiang Institute of Ecology and Geography, Chinese Academy of Sciences. A voucher specimen was deposited in the Natural Museum, **njiang Institute of Ecology and Geography, Chinese Academy of Sciences. (MS-20160919-015). M. sieversii seedlings were planted in 1/2 Murashige and Skoog (MS) medium with 30 g·L− 1 sucrose and 6 g·L− 1 agar in a greenhouse with 32 μmol·m− 2 s− 1 light intensity, 40–50% relative humidity, and a 16 h light/8 h dark photoperiod at 24 °C. Eight-week-old well-developed wild apples grew in medium at 4 °C under a 16 h photoperiod for two days (cold acclimation). Then, the chamber was cooled gradually (2 °C/h) from 4 °C to − 4 °C for 4 h (Boxun, LRX-580C-LED). Remove upper and lower three leaves and the middle leaves were separately harvested at the time points of CK, 0, 1, 6 and 10 h and each sample contained three biological replicates. Leaf samples were frozen in liquid nitrogen and stored at − 80 °C before physiological change analysis, hormone concentration determination, and RNA extraction. The RNA-seq was conducted using fifteen samples (CK, 0, 1, 6, 10 h) and the PacBio sequencing was implemented using the mixture of the samples.
Measurement of physiological changes and endogenous hormone contents
Before sequencing the transcriptome, we determined the physiological indicators. MDA was measured according to a previously published method [73]. Chlorophyll fluorescence was recorded with a PAM2500 fluorometer (Walz, Germany), and the maximal quantum efficiency of PSII was calculated as Fv/Fm (Fv = Fm-F0). REL was conducted using the procedure described by this method [74], and it was calculated based on the following equation:
The determination of endogenous ABA and JA content was performed using HPLC (Comin Suzhou, China), with three replicates for each analysis.
RNA quantification and transcriptome sequencing
Total RNA was isolated from the leaves of freeze-treated and untreated M. sieversii samples using the E.Z.N.A. Plant RNA Kit (Omega Bio-Tek, Norcross, GA, USA). Equal quantities of RNA from three biological replications at each stage were pooled to construct a complementary cDNA library. RNA concentration and quality were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). cDNA library construction was performed by high-throughput sequencing using the PacBio and Illumina platforms at Novogene (Bei**g, China). Additional nucleotide errors in consensus reads were corrected using the RNA-seq data with the software LoRDEC [75]. Index of the reference genome sequences was built using HISAT2 (v2.1.0) and paired-end clean reads were aligned to the reference genome sequences using HISAT2. Consensus reads were aligned to the reference genomic sequence (GDDH13, Version 1.0) using GMAP with parameters-no-chimeras-cross-species-expand-off sets1-B5-K50000-f samse-n1 against the reference genome sequences [76]. Gene structure analysis was performed using the TAPIS pipeline [77]. The GMAP output bam format file and gff/gtf format genome sequences annotation file were used for gene and transcript determination. Alternative splicing events and alternative polyadenylation events were then analyzed. Fusion transcripts were determined as transcripts mapped to two or more long-distance genes and were validated by at least two Illumina reads.
Iso-Seq data analysis
Raw reads were processed into error-corrected reads of interest (ROIs) according to the conditions full passes ≥0 and sequence accuracy> 0.75. By detecting whether each ROI sequence contains a 5′ primer, 3′ primer, and poly-A tail, the sequences can be divided into full-length sequences and non-full-length sequences. Based on the poly-A tail signal and the 5′ and 3′ cDNA primers in the ROIs, FLNC transcripts were identified [78]. The ICE (Isoform-clustering) algorithm in SMRT Analysis (v2.3.0) software was used to obtain a consistent sequence isoform, and the full-length consensus sequences were polished using Quiver. Full-length transcripts with a post correction accuracy above 99% were generated for further analysis. The redundant sequences were removed from the high-quality and corrected low-quality transcript sequences of each sample using CD-HIT (v4.6.8) software [79].
Differential expression analyses
Analysis of transcript expression levels used a fragments per kilobase of transcript per million mapped reads (FPKM) method. Differential expression analysis was performed using the DESeq R package (Version 1.18.0). The P values were adjusted using the Benjamini & Hochberg method. A corrected P-value of 0.05 and log2 (fold change) of 1 were set as the thresholds for significant differential expression.
GO enrichment analysis of DETs or lncRNA target genes was performed by the GO seq R package [80], in which gene length bias was corrected. GO terms with corrected P-values less than 0.05 were considered significantly enriched for differentially expressed genes. KEGG is a database resource for understanding the high-level functions and utilities of biological systems, such as cells, organisms, and ecosystems [81], from molecular level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (http://www.genome.jp/kegg/). KOBAS (v.3.0) software was used to test the statistical enrichment of DETs in KEGG pathways [82]. Plant transcription factors were predicted using iTAK (v1.7A) software [83].
Quantitative real-time PCR analysis of DETs
The qRT-PCR assays were performed to validate the consistency of RNA-Seq analysis. QRT-PCR analysis was performed using a Light Cycler 480 machine with SYBR Green I Master Mix (TaKaRa, China). Three biological replicates were set for each treatment. The relative expression of each transcript was calculated after normalization to the reference gene. The relative quantification from three biological replications was normalized to the reference gene and was calculated by the 2-ΔΔCt method. All primer sequences are shown in Additional file 15. Three technical replicates were performed based on the 2-ΔΔCt algorithm using EF1α as a quantitative control.
Statistical analysis
Statistical analysis was performed using ANOVA, and then a new multirange test was performed using Duncan’s range test in SPSS version 16.0. A significance level of P < 0.05 was applied to indicate significant values. Three independent biological replicates were used for RNA-seq and physiological and hormonal assays. Graphs were created using Origin 8.0 (Version 9.1; Origin Lab, Northampton, MA, USA).
Availability of data and materials
The sequence data have been deposited in the NCBI under BioProject accession number PRJNA706196 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA706196?reviewer=dm9b28i3sjt7q3m8mngn3npnal).
Abbreviations
- CA:
-
Cold acclimation
- DEG:
-
Differentially expressed gene
- JA:
-
Jasmonic acid
- ABA:
-
Abscisic acid
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- FPKM:
-
Fragments per kilobase of transcript per million mapped reads
- FLNC:
-
Full-length non-chimeric reads
- Iso-Seq:
-
Isoform Sequencing
- CCS:
-
Circular consensus sequence
- REC:
-
Relative electrolyte leakage values
- Fv/Fm:
-
Maximum photosynthetic efficiency values
- GMAP:
-
Genomic Map** and Alignment Program
- AS:
-
Alternative splicing
- DETs:
-
Differentially expressed transcripts
References
Harshman JM, Evans KM, Allen H, Potts R, Flamenco J, Aldwinckle HS, et al. Fire blight resistance in wild accessions·of Malus sieversii. Plant Dis. 2017;101(10):1738–45. https://doi.org/10.1094/PDIS-01-17-0077-RE.
Yan G, Long H, Song W, Chen R. Genetic polymorphism of Malus sieversii populations in **njiang. China Genetic Res Crop Evol. 2008;55(1):171–81. https://doi.org/10.1007/s10722-007-9226-5.
Wang Z-H, et al. Characterization of CBF1, CBF2, CBF3, and CBF4 genes of Malus sieversii and analysis of their expression in different habitats. Eur J Hortic Sci. 2017;82(2):81–9. https://doi.org/10.17660/eJHS.2017/82.2.3.
Banerjee A, et al. Epigenetic control of plant cold responses. Front Plant Sci. 2017;8.
Levitt J. 1980. Responses of plants to environmental stresses. New York: academic. 2nd ed.
Thomashow MF. Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu Rev Biochem. 1999;50(1):571–99. https://doi.org/10.1146/annurev.arplant.50.1.571.
Levitt J. Responses of plants to environmental stresses. Volume I. chilling, freezing, and high temperature stresses. In: Kozlowski TT, editor. Physiological ecology. New York: Academic Press; 1980. p. 497.
Steponkus PL, Uemura M, Webb MS. Membrane destabilization during freeze-induced dehydration. Curr Topics Plant Physiol. 1993;10:37–47.
Stockinger EJ, Gilmour SJ, Thomashow MF. Arabidopsis thaliana CBF1 encodes an AP2 domain-containing transcriptional activator that binds to the C-repeat/DRE, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc Natl Acad Sci U S A. 1997;94(3):1035–40. https://doi.org/10.1073/pnas.94.3.1035.
Feng, X.-M., et al. (2012). "the cold-induced basic helix-loop-helix transcription factor gene MdCIbHLH1 encodes an ICE-like protein in apple." BMC Plant biology 12.
An J-P, Wang XF, Zhang XW, Xu HF, Bi SQ, You CX, et al. An apple MYB transcription factor regulates cold tolerance and anthocyanin accumulation and undergoes MIEL1-mediated degradation. Plant Biotechnol J. 2020;18(2):337–53. https://doi.org/10.1111/pbi.13201.
Thomashow MF. Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu Rev Plant Physiol Plant Mol Biol. 1999;50(1):571–99. https://doi.org/10.1146/annurev.arplant.50.1.571.
Pagter M, Arora R. Winter survival and deacclimation of perennials under warmer climate: physiological perspectives. Physiol Plant. 2013;147(1):75–87. https://doi.org/10.1111/j.1399-3054.2012.01650.x.
Fitter AH, Fitter RSR. Rapid changes in flowering time in British plants. Science. 2002;296(5573):1689–91. https://doi.org/10.1126/science.1071617.
Dunn MA, Goddard NJ, Zhang L, Pearce RS, Hughes MA. Low-temperature-responsive barley genes have different control mechanisms. Plant Mol Biol. 1994;24(6):879–88. https://doi.org/10.1007/BF00014442.
Fowler S, Thomashow MF. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell. 2002;14(8):1675–90. https://doi.org/10.1105/tpc.003483.
Roman A, Andreu V, Hernandez ML, et al. Contribution of the difffferent omega-3 fatty acid desaturase genes to the cold response in soybean. J Exp Bot. 2012;63(13):4973–82. https://doi.org/10.1093/jxb/ers174.
Zheng C, Zhao L, Wang Y, Shen J, Zhang Y, Jia S, et al. Integrated RNA-seq and sRNA-seq analysis identifies chilling and freezing responsive key molecular players and pathways in tea plant (Camellia sinensis). PLoS One. 2015;10(4):e0125031. https://doi.org/10.1371/journal.pone.0125031.
Zhang Y, Zhang Y, Lin Y, et al. A Transcriptomic Analysis Reveals Diverse Regulatory Networks That Respond to Cold Stress in Strawberry (Fragaria×ananassa). International Journal Of Genomics. 2019;2019: Article 7106092.
Liang Y, Wang S, Zhao C, Ma X, Zhao Y, Shao J, et al. Transcriptional regulation of bark freezing tolerance in apple (Malus domestica Borkh.). Horticulture Research. 2020;7(1):205. https://doi.org/10.1038/s41438-020-00432-8.
**n H, Ni X, Pan X, et al. Comparative Transcriptome Analyses Revealed Conserved and Novel Responses to Cold and Freezing Stress in Brassica napus L. G3-Genes Genomes Genetics. 2019;9(8):2723–2737.
Deng S, Ma J, Zhang L, Chen F, Sang Z, Jia Z, et al. De novo transcriptome sequencing and gene expression profiling of Magnolia wufengensis in response to cold stress. BMC Plant Biol. 2019;19(1):321. https://doi.org/10.1186/s12870-019-1933-5.
Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20(11):631–56. https://doi.org/10.1038/s41576-019-0150-2.
Wang XC, Zhao QY, Ma CL, Zhang ZH, Cao HL, Kong YM, et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genomics. 2013;14(1):415. https://doi.org/10.1186/1471-2164-14-415.
Swindell WR, Huebner M, Weber AP. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics. 2007;8(1):125. https://doi.org/10.1186/1471-2164-8-125.
Li XL, et al. Transcriptome analysis provides insights into the stress response crosstalk in apple (Malus x domestica) subjected to drought, cold and high salinity. Sci Rep. 2019;9:10.
Sun M, et al. Transcriptome analysis of heat stress and drought stress in pearl millet based on Pacbio full-length transcriptome sequencing. BMC Plant Biol. 2020;20(1):15.
Wang S, Liang D, Li C, Hao Y, Ma F, Shu H. Influence of drought stress on the cellular ultrastructure and antioxidant system in leaves of drought-tolerant and drought-sensitive apple rootstocks. Plant Physiol Biochem. 2012;51:81–9. https://doi.org/10.1016/j.plaphy.2011.10.014.
Gregory PJ, Atkinson CJ, Bengough AG, Else MA, Fernández-Fernández F, Harrison RJ, et al. Contributions of roots and rootstocks to sustainable, intensified crop production. J Exp Bot. 2013;64(5):1209–22. https://doi.org/10.1093/jxb/ers385.
Welling A, Palva ET. Molecular control of cold acclimation in trees. Physiol Plantarum. 2006;127(2):167–81. https://doi.org/10.1111/j.1399-3054.2006.00672.x.
Hao XY, et al. Comprehensive transcriptome analysis reveals common and specific genes and pathways involved in cold acclimation and cold stress in tea plant leaves. Sci Hortic. 2018;240:354–68. https://doi.org/10.1016/j.scienta.2018.06.008.
Kaplan F, Sung DY, Guy CL. Roles of beta-amylase and starch breakdown during temperatures stress. Physiol Plant. 2006;126(1):120–8. https://doi.org/10.1111/j.1399-3054.2006.00604.x.
Sicher R. Carbon partitioning and the impact of starch deficiency on the initial response of Arabidopsis to chilling temperatures. Plant Sci. 2011;181(2):167–76. https://doi.org/10.1016/j.plantsci.2011.05.005.
Zhao L, Yang T, **ng C, et al. The beta-amylase PbrBAM3 from pear (Pyrus betulaefolia) regulates soluble sugar accumulation and ROS homeostasis in response to cold stress. Plant science. 2019;287: Article 110184.
Storm AR, Kohler MR, Berndsen CE, Monroe JD. Glutathionylation inhibits the catalytic activity of Arabidopsis β-Amylase3 but not that of paralog β-Amylase1. Biochemistry. 2018;57(5):711–21. https://doi.org/10.1021/acs.biochem.7b01274.
Yamdeu JHG, Gupta PH, Shah AK, et al. Profiling of StvacINV1, BAM1 and INH2 alpha expressions in relation to acid Invertase and beta-amylase activities during development of cold-induced sweetening in Indian potato (Solanum tuberosum L.) tubers. Am J Potato Res. 2015;92(5):603–8. https://doi.org/10.1007/s12230-015-9473-5.
Strauss G, Hauser H. STABILIZATION OF LIPID BILAYER VESICLES BY SUCROSE DURING FREEZING. Proc Natl Acad Sci U S A. 1986;83(8):2422–6. https://doi.org/10.1073/pnas.83.8.2422.
Wang Z, Wei P, Wu M, Xu Y, Li F, Luo Z, et al. Analysis of the sucrose synthase gene family in tobacco: structure, phylogeny, and expression patterns. Planta. 2015;242(1):153–66. https://doi.org/10.1007/s00425-015-2297-1.
Mittler R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002;7(9):405–10. https://doi.org/10.1016/S1360-1385(02)02312-9.
Demidchik V, Straltsova D, Medvedev SS, Pozhvanov GA, Sokolik A, Yurin V. Stress-induced electrolyte leakage: the role of K-permeable channels and involvement in programmed cell death and metabolic adjustment. J Exp Bot. 2014;65(5):1259–70. https://doi.org/10.1093/jxb/eru004.
Tsikas D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Anal Biochem. 2017;524:13–30. https://doi.org/10.1016/j.ab.2016.10.021.
Zhang Q, Kong X, Yu Q, et al. Responses of PYR/PYL/RCAR ABA Receptors to Contrasting stresses, Heat and Cold in Arabidopsis. Plant signaling & behavior. 2019;14(12): Article 1670596.
Zhang Y, Shi SH, Li FL, Zhao CZ, Li AQ, Hou L, et al. Global transcriptome analysis provides new insights in Thellungiella salsuginea stress response. Plant Biol. 2019;21(5):796–804. https://doi.org/10.1111/plb.13006.
Seki M, Umezawa T, Urano K, Shinozaki K. Regulatory metabolic networks in drought stress responses. Curr Opin Plant Biol. 2007;10(3):296–302. https://doi.org/10.1016/j.pbi.2007.04.014.
Gusta LV, Trischuk R, Weiser CJ. Plant cold acclimation: the role of abscisic acid. J Plant Growth Regul. 2005;24(4):308–18. https://doi.org/10.1007/s00344-005-0079-x.
Li CY, Puhakainen T, Welling A, Viherä-Aarnio A, Ernstsen A, Junttila O, et al. Cold acclimation in silver birch (Betula pendula). Development of freezing tolerance in different tissues and climatic ecotypes. Physiol Plantarum. 2002;116(4):478–88. https://doi.org/10.1034/j.1399-3054.2002.1160406.x.
Carvajal F, Palma F, Jimenez-Munoz R, et al. Unravelling the role of abscisic acid in chilling tolerance of zucchini during postharvest cold storage. Postharvest Biol Technol. 2017;133:26–35. https://doi.org/10.1016/j.postharvbio.2017.07.004.
Fuchs S, Tischer SV, Wunschel C, Christmann A, Grill E. Abscisic acid sensor RCAR7/PYL13, specific regulator of protein phosphatase coreceptors. Proc Natl Acad Sci U S A. 2014;111(15):5741–6. https://doi.org/10.1073/pnas.1322085111.
Fujita Y, Fujita M, Satoh R, Maruyama K, Parvez MM, Seki M, et al. AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell. 2005;17(12):3470–88. https://doi.org/10.1105/tpc.105.035659.
Lee SJ, Kang JY, Park HJ, Kim MD, Bae MS, Choi HI, et al. DREB2C interacts with ABF2, a bZIP protein regulating abscisic acid-responsive gene expression, and its overexpression affects abscisic acid sensitivity. Plant Physiol. 2010;153(2):716–27. https://doi.org/10.1104/pp.110.154617.
Jasmonate regulates the inducer of cbf expression-C-repeat binding factor/DRE binding factor1 cascade and freezing tolerance in Arabidopsis. Plant Cell25:2907–2924.
Hu YR, et al. Jasmonate regulates leaf senescence and tolerance to cold stress: crosstalk with other phytohormones. J Exp Bot. 2017;68(6):1361–9. https://doi.org/10.1093/jxb/erx004.
Bari R, Jones JDG. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69(4):473–88. https://doi.org/10.1007/s11103-008-9435-0.
Yu XM, Griffith M, Wiseman SB. Ethylene induces antifreeze activity in winter rye leaves. Plant Physiol. 2001;126(3):1232–40. https://doi.org/10.1104/pp.126.3.1232.
Guo HW, Ecker JR. The ethylene signaling pathway: new insights. Curr Opin Plant Biol. 2004;7(1):40–9. https://doi.org/10.1016/j.pbi.2003.11.011.
Shibasaki K, Uemura M, Tsurumi S, et al. Auxin response in Arabidopsis under cold stress: underlying molecular mechanisms. Plant Cell. 2009;21:3823–38.
Miura K, Lee J, Gong Q, et al. SIZ1 regulation of phosphate starvation-induced root architecture remodeling involves the control of auxin accumulation. Plant Physiol. 2011;155:1000–12.
Guan S, Xu Q, Ma D, Zhang W, Xu Z, Zhao M, et al. Transcriptomics profiling in response to cold stress in cultivated rice and weedy rice. Gene. 2009;685:96–105. https://doi.org/10.1016/j.gene.2018.10.066.
Nouri MZ, Moumeni A, Komatsu S. Abiotic stresses: insight into gene regulation and protein expression in photosynthetic pathways of plants. Int J Mol Sci. 2015;16(9):20392–416. https://doi.org/10.3390/ijms160920392.
Gururani MA, Venkatesh J, Lam-Son PT. Regulation of photosynthesis during abiotic stress-induced Photoinhibition. Mol Plant. 2015;8(9):1304–20. https://doi.org/10.1016/j.molp.2015.05.005.
Salazar J, et al. Transcriptome analysis and postharvest behavior of the kiwifruit 'Actinidia deliciosa' reveal the role of ethylene-related phytohormones during fruit ripening. Tree Genet Genomes. 2021;17(1):19.
Lindemose S, O’Shea C, Jensen MK, Skriver K. Structure, function and networks of transcription factors involved in abiotic stress responses. Int J Mol Sci. 2013;14(3):5842–78. https://doi.org/10.3390/ijms14035842.
Gong X-X, Yan BY, Hu J, Yang CP, Li YJ, Liu JP, et al. Transcriptome profiling of rubber tree (Hevea brasiliensis) discovers candidate regulators of the cold stress response. Genes & Genomics. 2018;40(11):1181–97. https://doi.org/10.1007/s13258-018-0681-5.
Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. AP2/ERF family transcription factors in plant abiotic stress responses. BBA-Gene Regul Mech. 2012;1819(2):86–96. https://doi.org/10.1016/j.bbagrm.2011.08.004.
**e Y, Chen P, Yan Y, Bao C, Li X, Wang L, et al. An atypical R2R3 MYB transcription factor increases cold hardiness by CBF-dependent and CBF-independent pathways in apple. New Phytol. 2018;218(1):201–18. https://doi.org/10.1111/nph.14952.
**e XB, et al. The bHLH transcription factor MdbHLH3 promotes anthocyanin accumulation and fruit colouration in response to low temperature in apples. Plant Cell and Environment. 2012;35(11):1884–97. https://doi.org/10.1111/j.1365-3040.2012.02523.x.
Song TT, et al. Identification of new regulators through transcriptome analysis that regulate anthocyanin biosynthesis in apple leaves at low temperatures. PLoS One. 2019;14(1):19.
Yao GF, et al. Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017;92(3):437–51. https://doi.org/10.1111/tpj.13666.
Wu R, Wang Y, Wu T, Xu X, Han Z. MdMYB4, an R2R3-type MYB transcription factor, plays a crucial role in cold and salt stress in apple calli. J Am Soc Hortic Sci. 2017;142(3):209–16. https://doi.org/10.21273/JASHS04030-17.
Chen JN, et al. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in Brassinosteroid-regulated plant growth and drought responses. Plant Cell. 2017;29(6):1425–39. https://doi.org/10.1105/tpc.17.00364.
Yu YC, et al. WRKY71 acts antagonistically against salt-delayed flowering in Arabidopsis thaliana. Plant Cell Physiol. 2018;59(2):414–22. https://doi.org/10.1093/pcp/pcx201.
Liu X, Li X, Wen X, Zhang Y, Ding Y, Zhang Y, et al. PacBio full-length transcriptome of wild apple (Malus sieversii) provides insights into canker disease dynamic response. BMC Genomics. 2021;22(1):52. https://doi.org/10.1186/s12864-021-07366-y.
Dhindsa RS, Plumb-Dhindsa P, Thorpe TA. Leaf senescence: correlated with increased levels of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J Exp Bot. 1981;32(1):93–10. https://doi.org/10.1093/jxb/32.1.93.
Bajji M, Kinet JM, Lutts S. The use of the electrolyte leakage method for assessing cell membrane stability as a water stress tolerance test in durum wheat. Plant Growth Regul. 2002;36(1):61–70. https://doi.org/10.1023/A:1014732714549.
Salmela L, Rivals E. LoRDEC: accurate and efficient long read error correction. Bioinformatics. 2014;30(24):3506–14. https://doi.org/10.1093/bioinformatics/btu538.
Wu TD, Watanabe CK. GMAP: a genomic map** and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21(9):1859–75. https://doi.org/10.1093/bioinformatics/bti310.
Abdel-Ghany SE, Hamilton M, Jacobi JL, Ngam P, Devitt N, Schilkey F, et al. A survey of the sorghum transcriptome using single-molecule long reads. Nat Commun. 2016;7(1):11706. https://doi.org/10.1038/ncomms11706.
Hou C, Deng N, Su YJF. PacBio long-read sequencing reveals the transcriptomic complexity and aux/IAA gene evolution in Gnetum (Gnetales). Forests. 2019;10(11):1043. https://doi.org/10.3390/f10111043.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–315. https://doi.org/10.1093/bioinformatics/bts565.
Core R. Team. R: a language and environment for statistical computing version 3.2.0. Vienna: R Foundation for Statistical. Computing. 2018.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51. https://doi.org/10.1002/pro.3715.
**e, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotationand identification of enriched pathways and diseases. Nucleic Acids Res.2011,39, W316–W322.
Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P, et al. iTAK: a programfor genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant. 2016;9(12):1667–70. https://doi.org/10.1016/j.molp.2016.09.014.
Acknowledgments
We acknowledge the Novogene Corporation (Bei**g, China) for the facilities and expertise of the PacBio platform for libraries construction, sequencing, and bioinformatics analysis.
Funding
This work was supported by the Second Tibetan Plateau Scientific Expedition.
and Research (STEP) program (2019QZKK0502030403), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (No.2018478), and NSFC-XJ key project (No. U1903206).
Author information
Authors and Affiliations
Contributions
PZ, XJW, XSL, and DYZ conceived the study, PZ carried out the experiments, XJW and XSL provided advice, XJL and YZ participated in data interpretation. DYZ revised the article. All authors approved the final draft of the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The seeds of M. sieversii collection were performed under institutional guidelines under local legislation. Seeds of M. sieversii were collected in September 2016 from one area (43°23′2.20″N; 83°35′43.48″E) in a natural wild reserve forest in Yili, **njiang. With the permission of the **nyuan County Forestry Bureau, seeds were obtained from the field and identified. The voucher specimen is stored in the Natural Museum of **njiang Institute of ecological geography, Chinese Academy of Sciences (MS-20160919-015). The germplasm of M. sieversii was identified by Ph.D. Wenjun Li, who worked at the **njiang Institute of ecological geography, Chinese Academy of Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Characterization of M. sieversii transcripts. (A) The number of consensus reads in different lengths; (B) Novel gene statistical chart of annotation results of seven databases (NR, SwissProt, KEGG, KOG/COG, GO, NT, PFAM).
Additional file 2: Fig. S2.
GO and KEGG enrichment analysis for 2234 DEGs. A: GO enrichment analysis for 2234 DEGs. B: KEGG enrichment analysis for 2234 DEGs.
Additional file 3: Fig. S3.
CIRCOS visualization. The order of Circos from outside to inside are: a. Chromosomes of M.domestica. b. AS position. (Stacking histogram, turquoise: RI; green: A3; yellow: A5; purple: SE; red: MX; brown: AF; dark blue: AL). c. APA position mapped to chromosomes. d. Novel transcript density. e. Novel gene density. f. LncRNA density distribution. g. Fusion transcript distribution. Intrachromosome (purple); inter-chromosome (yellow).
Additional file 4: Fig. S4.
Analysis of differentially expressed transcripts (DETs). (A) KEGG pathway enrichment on 1 h vs CK. (B) KEGG pathway enrichment on 6 h vs CK. (C) KEGG pathway enrichment on common DETs at different time points.
Additional file 5: Fig. S5.
Validation of expression pattern of DEGs by qRT-PCR. Expression levels were analyzed using a T-test. Within each figure, asterisks above the bars indicate statistical significance (*p < 0.05; **p < 0.01). The normalized expression level (FPKM) of Illumina sequencing is indicated on the left y-axis and the relative expression of qRT-PCR is indicated on the right y-axis. Three biological replicates were set for each treatment. Experiments were repeated at least three times with similar results.
Additional file 6: Table S1.
Details regarding AS.
Additional file 7: Table S2.
Details regarding fusion gene.
Additional file 8: Table S3.
Details regarding all DETs.
Additional file 9: Table S4.
List of 2234 DEGs.
Additional file 10: Table S5.
List of 4410 DETs.
Additional file 11: Table S6.
GO enrichment analysis for 4410 DETs.
Additional file 12: Table S7.
KEGG enrichment analysis for 4410 DETs.
Additional file 13: Table S8.
Details regarding pathways.
Additional file 14: Table S9.
Details regarding TFs.
Additional file 15: Table S10.
Primers used for RT-qPCR verification.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhou, P., Li, X., Liu, X. et al. Transcriptome profiling of Malus sieversii under freezing stress after being cold-acclimated. BMC Genomics 22, 681 (2021). https://doi.org/10.1186/s12864-021-07998-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07998-0