
Abstract
Background
To facilitate the clinical implementation of genomic medicine by next-generation sequencing, it will be critically important to obtain accurate and consistent variant calls on personal genomes. Multiple software tools for variant calling are available, but it is unclear how comparable these tools are or what their relative merits in real-world scenarios might be.
Methods
We sequenced 15 exomes from four families using commercial kits (Illumina HiSeq 2000 platform and Agilent SureSelect version 2 capture kit), with approximately 120X mean coverage. We analyzed the raw data using near-default parameters with five different alignment and variant-calling pipelines (SOAP, BWA-GATK, BWA-SNVer, GNUMAP, and BWA-SAMtools). We additionally sequenced a single whole genome using the sequencing and analysis pipeline from Complete Genomics (CG), with 95% of the exome region being covered by 20 or more reads per base. Finally, we validated 919 single-nucleotide variations (SNVs) and 841 insertions and deletions (indels), including similar fractions of GATK-only, SOAP-only, and shared calls, on the MiSeq platform by amplicon sequencing with approximately 5000X mean coverage.
Results
SNV concordance between five Illumina pipelines across all 15 exomes was 57.4%, while 0.5 to 5.1% of variants were called as unique to each pipeline. Indel concordance was only 26.8% between three indel-calling pipelines, even after left-normalizing and intervalizing genomic coordinates by 20 base pairs. There were 11% of CG variants falling within targeted regions in exome sequencing that were not called by any of the Illumina-based exome analysis pipelines. Based on targeted amplicon sequencing on the MiSeq platform, 97.1%, 60.2%, and 99.1% of the GATK-only, SOAP-only and shared SNVs could be validated, but only 54.0%, 44.6%, and 78.1% of the GATK-only, SOAP-only and shared indels could be validated. Additionally, our analysis of two families (one with four individuals and the other with seven), demonstrated additional accuracy gained in variant discovery by having access to genetic data from a multi-generational family.
Conclusions
Our results suggest that more caution should be exercised in genomic medicine settings when analyzing individual genomes, including interpreting positive and negative findings with scrutiny, especially for indels. We advocate for renewed collection and sequencing of multi-generational families to increase the overall accuracy of whole genomes.
Similar content being viewed by others

Background
Recent studies have substantiated the prevalence of rare mutations in the human genome [1, 2]. Whole-genome sequencing (WGS) can uncover substantially more genetic variation than traditional single-nucleotide polymorphism (SNP) arrays, thus explaining a larger fraction of human phenotypic diversity [3, 4]. This in turn is driving the sequencing of personal genomes aimed at obtaining highly accurate information about each person's genome [5, 6].
Given the existence of multiple sequencing platforms and multiple data-analysis pipelines for next-generation sequencing, researchers and clinicians may be under the impression that these methods all work similarly to identify genetic variants from personal genomes. However, one group recently reported that when variants detected in the same sample by the 1000 genomes project (1 KGP) and the Complete Genomics (CG) platform were compared, 19% of the single-nucleotide variants (SNVs) derived were unique to one dataset [7]. This is likely due to differences in technology, data collection, read-alignment methods, and variant-calling algorithms. The group further concluded that 'current research resources and informatics methods do not adequately account for the high level of variation that already exists in the human population, and significant efforts are needed to create resources that can accurately assess personal genomes for health, disease, and prediction of treatment outcomes' [7]. As an illustration of the widely differing methods currently being used, one of the above-referenced papers used Illumina sequencing data processed with the Short Oligonucleotide Analysis Package (SOAP) pipeline [2] whereas the other group used Illumina sequencing data processed with the Genome Analysis Toolkit (GATK) pipeline [1]. Neither group published a comparison of the overlap (concordance or discordance) between pipelines. Other researchers have worked on establishing a rigorous filtering pipeline to optimize SNV calling, reporting that the cumulative application of 12 individual filters resulted in a 290-fold reduction in the error rate [8]. Another group has worked to optimize their own pipeline utilizing, among other things, GATK and SAMtools, although it is not clear if this group compared their results with anything from SOAP [9]. This same group published a comparison of data obtained using sequencing from Illumina and CG, which showed an unexpectedly high level of discordance between the two platforms [10], which has been debated in blog postings [11, 12].
Despite these previous studies comparing technical platforms, there have not been many published systematic evaluations of a number of currently used bioinformatics pipelines when generating variant calls from the same set of raw sequence data. Additionally, despite the existence of many variant-calling software tools [13], their concordance using near-default settings has not been thoroughly investigated, making it difficult to assess the relative effects on variant calling of differences in sequencing platforms versus differences in implementations of bioinformatics pipelines. Ideally, researchers and clinicians should have little to no uncertainty about the correct pipeline parameterizations for each sequencing experiment, and hence little variability with respect to their pipeline implementations; however this is rarely, if ever, the case. Indeed, knowledge about the perfect and most appropriate parameterization is often not available or easily obtainable when performing in-depth sequence analysis, and, sometimes the 'correct' parameters may never be precisely characterized due to the complex nature of the experiment. Researchers, clinicians and policy-makers stand to benefit from a greater understanding of the variability introduced by imperfect and non-standardized implementations of the available bioinformatics pipelines.
To address this issue, we carried out a study of 15 exomes and one whole genome from 15 research participants, analyzing the data with a range of different variant-calling pipelines using near-default parameters. Our results have significant implications for analyzing personal genomes from next-generation sequencing experiments.
Methods
Ethics approval
The collection and genomic analysis of the DNA were approved by the institutional review board at the University of Utah, and written informed consent was obtained from all study participants. Research was carried out in compliance with the Helsinki Declaration.
Sample collection
The samples used in our study all came from families of human research participants ascertained in clinics at the University of Utah (see Additional file 1, Figure S1 for pedigrees). Blood samples were collected and genomic DNA extracted using alkaline lysis and ethanol precipitation (Gentra Puregene; Qiagen Corp., Valencia, CA USA). DNA was quality-checked on agarose gels and quantified using a microvolume spectrophotometer (NanoDrop 2000; Thermo Fisher Scientific Inc., West Palm Beach, FL, USA).
Whole-genome sequencing and analysis with Complete Genomics
After quality control to ensure lack of genomic degradation, we sent DNA samples (10 ug) to Complete Genomics (CG) (Mountain View, CA, USA) for sequencing. The whole-genome DNA was sequenced using nanoarray-based short-read sequencing by ligation technology [14], including an adaptation of the pairwise end-sequencing strategy [15]. Reads were mapped to the Genome Reference Consortium assembly GRCh37. Owing to the proprietary data formats, all the sequencing data quality control, alignment, and variant calling were performed by CG as part of their sequencing service, using their version 2.0 pipeline [16].
Exome capture and sequencing with Illumina HiSeq2000
Exome capture for all 15 samples was carried out using a commercially available in-solution method (SureSelect Human All Exon v2; Agilent Technologies Inc., Wilmington, DE, USA), following the manufacturer's guidelines. This method is designed to target all human exons, regions totaling approximately 44 Mb, covering 98.2% of the Consensus Coding Sequence (CCDS) database. For the capture, a DNA-shearing instrument (focused-ultrasonicator; Covaris Inc., Woburn, MA, USA) was used to randomly fragment the pure and high molecular weight genomic DNA samples (experiments carried out by BGI-Shenzhen, Shenzhen, China), resulting in DNA fragments with a base-pair peak of 150 to 200 bp. Adaptors were then ligated to both ends of the resulting fragments. The adaptor-ligated templates were purified by magnetic beads (Agencourt AMPure SPRI; Beckman Coulter Inc., Brea, CA, USA), and fragments with an insert size of approximately 250 bp were excised. Extracted DNA was amplified by ligation-mediated (LM)-PCR, purified, and hybridized (SureSelect Library; Agilent Technologies) for enrichment. Hybridized fragments bound to the strepavidin beads, whereas the unbound non-hybridized fragments were washed out after 24 hours of hybridization. Captured LM-PCR products were analyzed using a microfluidics-based platform (2100 Bioanalyzer; Agilent Technologies) to estimate the magnitude of the enrichment. Paired-end sequencing was performed using a sequencing platform (HiSeq2000; Illumina Inc., San Diego, CA, USA) with average read lengths of 90 bp. Raw image files were processed (Pipeline version 1.6; Illumina Inc.) for base-calling, using the default parameters. FASTQ files were produced from the pipeline for downstream sequence data analysis. A gender check was compatible with the known genders of the collected human participants.
SNP arrays
DNA samples were genotyped on the SNP arrays (Human610-Quad, version 1; Illumina Inc.) with approximately 610,000 markers (including approximately 20,000 non-polymorphic markers) at the Center for Applied Genomics (Children's Hospital of Philadelphia, Philadelphia, PA USA). Total genomic DNA extracted from whole blood was used in the experiments. Standard data-normalization procedures and canonical genotype-clustering files provided by Illumina were used to process the genoty** signals. Concordance between SNPs from the arrays and SNPs from exome sequencing was determined by calculating the percentage of variants from exome sequencing and comparing this with the same genotype derived from the SNP arrays.
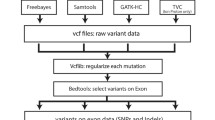
Alignment and variant calling
BWA-GATK variant calling
Burrows-Wheeler aligner (BWA; version 0.5.9 [17]) was used to align the sequencing reads, with default parameters, to the human reference genome sequence GRCh37. Alignments were converted from sequence alignment map (SAM) format to sorted, indexed binary alignment map (BAM) files (SAMtools version 0.1.18; http://sourceforge.net). The Picard tool was used to remove duplicate reads. GATK software tools (version 1.5; http://www.broadinstitute.org) were used for improvement of alignments and genotype calling and refining with recommended parameters [18]. BAM files were re-aligned with the GATK IndelRealigner, and base quality scores were re-calibrated by the GATK base quality recalibration tool. Genotypes were called by the GATK UnifiedGenotyper, and the GATK VariantRecalibrator tool was used to score variant calls by a machine-learning algorithm and to identify a set of high-quality SNPs using the Variant Quality Score Recalibration (VQSR) procedure. GATK was used to filter high-quality insertions and deletions (indels) by hard criteria, 'QD < 2.0, ReadPosRankSum < -20.0 FS > 200.0'. Finally, we removed SNVs and indels located outside of regions targeted by exome capture. To increase sensitivity, only those indels with depth (DP) of 10 or more, and with more than 4 reads supporting the indel events were included in the final high-confidence indel set. At a later date, one exome was processed with newer versions of the GATK v2.3-9 UnifiedGenotyper and GATK v2.3-9 HaplotypeCaller modules.
BWA-SAMtools genotype calling
Using the above BAM files, we used SAMtools (version 0.1.18) to generate genotype calls [19]. The 'mpileup' command in SAMtools was used to identify SNPs and indels, and we removed variants with DP coverage less than 10, and variants located outside of exome-capture regions.
SOAP pipeline
Adaptor and low-quality sequences were removed before map**. Sequence reads identified from each individual were then aligned to human reference genome GRCh37 using SOAPaligner (version 2.21 [20]) with a maximum of five mismatches. Duplicate reads were removed. Consensus genotypes in target regions were called by SOAPsnp (version 1.03) [21] with recommended parameters. SNV results were filtered (Phred-like SNV quality ≥ 20, overall depth 8 to 500, copy number estimate < 2, and distance between two adjacent SNVs ≥ 5). For a heterozygous SNV, the quality of the minor allele was required to be at least 20, depth of coverage for the minor allele at least 4, and the ratio of major allele to minor allele less than 5. For indel calling, SOAPindel was used, which adopts local assembly based on an extended de Bruijn graph [22]. For SOAPindel, the aligner BWA was used to align the reads to the human reference sequence with default parameters. Initially, putative indels were assumed to be located near the unmapped reads whose mates mapped to the reference genome. SOAPindel then executed a local assembly (k-mer = 25) on the clusters of unmapped reads. Clusters with coverage of less than 5 were not used. The assembly results were aligned to the reference in order to find the potential indels. To distinguish true-positive and false-positive indels, SOAPindel generates Phred quality scores, which take into consideration the depth of coverage, indel size, number of neighboring variants, distance to the edge of the contig, and position of the second different base pair. Only those indels with a quality score of 10 or higher were retained in the final indel call set.
GNUMAP pipeline
Diploid and monoploid SNVs for each individual were called using the GNUMAP pipeline (version 3.1.0 [23]). GNUMAP-SNP utilizes a novel probabilistic pair-hidden Markov model, which accounts for uncertainty in the read calls as well as read map** in an unbiased fashion. Raw reads were initially aligned to the full genome using an alignment score of 260 or greater, which for this dataset allowed for only one SNV per read. A k-mer size of 12 and a jump size of 10 were also used. Only SNVs within exome regions with a P < 0.001 were reported. The GNUMAP pipeline cannot currently call indels.
BWA-SNVer pipeline
BWA [17] was used to align the sequencing reads to GRCh37 with default parameters. Duplicate reads were removed by Picard, and SNVer (version 0.2.1) was then used for detecting SNVs in each sample [24]. Similar to GATK [18], only the mapped short reads with map** quality of greater than 20 were considered, and only bases with base quality greater than 17 counted. SNVer estimated the empirical error rate for those selected reads in making variant calls. We set the number of haploids to 2 for analysis of individual samples, and set the variant allele frequency threshold of greater than 0 for detecting both rare and common SNVs. SNVer provides multiplicity control, and we performed Bonferroni correction and controlled the family-wise error rate at the 0.05 level to report identified SNVs. Indels cannot currently be called by the BWA-SNVer pipeline.
Post-variant calling analyses
Post-variant-calling analyses were performed using Golden Helix SVS (version 7.6.10 [25], ANNOVAR [26], the R suite of statistical programming tools http://www.r-project.org, and custom Perl scripts.
MiSeq sequencing for validation
Validation variants were randomly selected from sets of particularly controversial variants, indels and SNVs unique to GATK, indels and SNVs unique to SOAP, and variants (both SNVs and indels) shared by these two pipelines. PCR primers were designed using the software program Primer 3 http://sourceforge.net, to produce amplicons (ranging in size from 100 to 200 bp) containing variants of interest in approximately the center of the amplicon. Primers were obtained in 96-well plate format, 10 μmol/L dilution each (Sigma-Aldrich, St Louis, MO, USA). All primers were first tested for PCR efficiency using a HAPMAP DNA sample (Catalog ID NA12864l Coriell Institute for Medical Research, Camden, NJ, USA) and DNA polymerase (LongAmp® Taq DNA Polymerase; New England Biolabs, Beverly, MA, USA). k8101-49685 genomic DNA was used as template for the validation experiment. After quality-control steps using agarose gel, the product was purified (ExoSAP-IT® reagentsl Affymetrix Inc., Santa Clara, CA, USA) and pooled. Final PCR products were quantified (Qubit® dsDNA BR Assay Kitl Invitrogen Corp., Carlsbad, CA, USA), then library construction for the sequencer platform (MiSeq Personal Sequencer; Illumina Inc.) was performed. Finally, before being loaded onto the MiSeq machine, the quality and quantity of the sample was verified using the Bioanalyzer (Agilent Technologies) and quantitative PCR (Kapa Biosystems Inc., Woburn, MA, USA).
Accessing data
All of the data have been submitted to the Sequence Read Archive under project accession SRP019719, corresponding to the 15 exomes and the single whole genome analyzed during the course of our study (see Additional file 2).
Results
Data production summary
Fifteen DNA samples from four different families (see Additional file 1, Figure S1) were prepared by exon capture (Agilent 44 MB SureSelect protocol; Agilent Technologies), followed by sequencing on (HiSeq2000; Illumina Inc.). On average, we obtained sequence coverage of approximately 120X (range, 100 to 154X) on targeted regions for these 15 samples. For all samples, sequence reads covered more than 80% of the targeted region with a depth of greater than 20 reads per base (see Additional file 1, Figure S2; for data production statistics, see Additional file 3). Five different pipelines were used for read alignment and variant calling (SNVs and indels when possible) (Table 1). In addition, one whole genome was sequenced and analyzed by CG with 95% of the exome region covered by 20 reads or more per base, resulting in greater than 88% of the genome covered with a depth of greater than 20 reads per base. Variant calls were generated by CG with their in-house analysis pipeline (version 2.0).
SNV analysis
Concordance with SNP genoty** arrays
Sensitivity and specificity for detecting common SNPs was calculated for each Illumina variant-calling pipeline for four samples that were genotyped with the Illumina Human610-Quad version 1 SNP array (see Additional file 1, Table S1). We caution that this analysis was restricted to a set of common SNPs targeted by the SNP array, and that these tend to be within regions containing little to no repeated sequences and without extreme GC contents. Therefore, although widely used in published literature, concordance with SNP arrays does not adequately measure real-world performance on all variants in personal genomes. With this major caveat in mind, performance for each pipeline was measured by treating the Illumina Human610-Quad version 1 SNP arrays as a true-positive reference, and comparing the exome-capture sequencing results with this reference set. The average specificity for each of the five Illumina pipelines was generally high, ranging from 99.59% to 99.87% (Table 2), consistent with the fact that each of these pipelines have been optimized to minimize false negatives for known common SNPs. The average sensitivity ranged among the five pipelines from 86.6% (with GNUMAP) to 95.3% (with GATK1.5). Sensitivity decreased when the variant set was iteratively restricted to the intersection between two or more variant-calling pipelines, whereas specificity naturally shows the opposite trend of increasing values under the same series of intersections (Table 2).
Evaluation of performance by inheritance analysis
To explore the validity of SNVs called by each Illumina pipeline, we performed an inheritance analysis for two families contained within the 15 sequenced exomes. Previous calculations have estimated the average expected number of de novo non-synonymous coding mutations per individual exome to be approximately 1 to 2 [27–30]. However, we found that the number of putative de novo mutations per child per exome was much higher if only the parents of the child were used to filter out inherited mutations. Adding an additional familial generation to the filtering process, in our case a grandparent, significantly reduced the number of putative de novo variants to a value comparable with that of the previously reported value of expected de novo non-synonymous mutations. In addition, significant variation was seen in the number of putative de novo mutations between the two families (Table 3), consistent with previous findings [31].
Variant-calling pipeline concordance
SNV concordance between all 5 Illumina pipelines across all 15 exomes was 57.4% on average, and Ti/Tv ratios showed a generally increasing trend for sets of variants intersected by an increasing number of variant-calling pipelines (Figure 1). We found that for novel SNVs (those not found in dbSNP135) the overall concordance (11.4%) was much lower than the overall concordance between known SNVs (59.6%) (Figure 1). In a previous paper, we validated with Sanger sequencing or Sequenom genoty** 17 SNVs found in 3 of the current pilot samples [33]. Of these 2,085 SNVs uniquely called by CG, an average of 558 had no sequence coverage as mapped by any of the Illumina-based exome-analysis pipelines. The Illumina exome read-depth for the remaining 1,527 CG-unique SNVs was calculated, and the majority of these SNVs were found to be in regions of very low Illumina sequence coverage (< 20 reads) in the exome datasets (see Additional file 1, Figure S5).

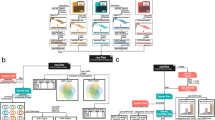
Single-nucleotide variant (SNV) concordance, between two sequencing pipelines (Illumina and Complete Genomics (CG)) for a single exome, k8101-49685. For the Illumina sequencing, exons were captured using the Agilent SureSelect version 2 panel of capture probes. CG SNVs consisted of a subset of all SNVs called by CG that fell within the Agilent SureSelect version 2 exons. Concordance was determined by matching the genomic coordinates, base-pair composition, and zygosity status for each detected SNVs. Illumina SNVs consisted of all SNVs (the union) called by the five variant-calling pipelines GATK, SAMtools, SOAPsnp, SNVer, and GNUMAP, which increased the false positives but decreased the false negatives. Concordance was measured between Illumina SNVs and (A) all CG SNVs, (C) only high-quality (VQHIGH) CG SNVs, and (D) only low quality (VQLOW) CG SNVs. (B) Genome mappability analyses were performed on 2,085 discordant SNVs, which were found by the CG pipeline and not found by any of the five Illumina data pipelines.
We found that 89.3% of CG SNVs (17,322 of 19,407) were contained within the union of all five Illumina pipelines (35,653 putative SNVs), whereas 18,331 of these 35,653 putative Illumina SNVs were not called by CG, suggesting a high false positive rate in the union of the Illumina calls and/or conversely a high false-negative rate in the CG calls (Figure 2). Overall concordance displayed marginal increases when VQLOW SNVs (low-quality CG variants) were removed from the pool of CG SNVs (Figure 2). Overall concordance remained stable as the depth of coverage threshold value associated with Illumina data calls increased (see Additional file 1, Figure S6A).
When only highly concordant Illumina SNVs (SNVs called by all five Illumina pipelines) were compared with the CG SNVs, only 64.4% (12507) of CG SNVs were contained within the concordant Illumina set, suggesting a high false-negative rate in this highly concordant Illumina set. Overall agreement decreased as the depth of coverage threshold value for Illumina calls increased, consistent with an increasing false-negative rate (see Additional file 1, Figure S6B).
Cross-platform comparison of unique-to-pipeline SNVs
SNVs from sample k8101-49685 that were uniquely detected by only one of the five Illumina variant-calling pipelines were compared with SNVs called by CG (see Additional file 1, Figure S7). Of the SNVs uniquely called by GATK, 809 of 1671 (48%) were concordant with CG data. The concordance was much lower for the other four pipelines, 49 of 1,102 SNVs (4%) for GNUMAP, 45 of 886 (5%) for SAMtools, 29 of 226 (12%) for SNVer, and 24 of 908 (3%) for SOAPsnp. Concordance improved for SNVs that were called by more than a single Illumina data pipeline, and the concordance was the highest for variants found by all five Illumina pipelines (see Additional file 1, Figure S7).
For variants that were novel as well as unique to a single Illumina pipeline, concordance with CG data was low (see Additional file 1, Figure S7). For GATK, 25% (13 of 51) of novel and unique-to-pipeline SNVs were concordant with CG data; for GNUMAP and SOAPsnp, no novel and unique-to-pipeline SNVs were concordant (0 of 470 and 0 of 229 respectively); for SAMtools, 0.2% (1 of 418) of novel and unique-to-pipeline SNVs were concordant; and for SNVer, 6% (1 of 18) of novel and unique-to-pipeline SNVs were concordant. Concordance rates of novel and unique-to-pipeline SNVs increased for variants called by an increasing number of pipelines (see Additional file 1, Figure S7).
Indel analysis
Variant-calling pipeline concordance
For indel calls, initial agreement between SOAPindel, SAMtools and GATK was very low at 3.0% (see Additional file 1, Figure S8). Indel coordinates were subsequently left-normalized and intervalized using a total range of 20 genomic coordinates (10 bp in each direction of their genomic coordinates). We found that increasing the intervalized indel range to as much as 60 genomic coordinates only marginally differed from having 20, so we chose to use 20 as a reasonable and conservative range for intervalizing indels. This method increased the overall concordance to 26.8% between the three indel-calling pipelines (Figure 3). For novel indels, the concordance (4.7%) was much lower than the overall concordance among known indels (43.3%). In an earlier paper, we previously validated with Sanger sequencing three indels found in three of the current pilot samples [Full size image
For indels found in sample k8101-49685, 960 were randomly selected for validation. Of these, 386 were randomly selected from the unique-to-GATK indel set, 387 were randomly selected from the unique-to-SOAPindel set, and 187 were randomly selected from set of indels overlap** between the two (SOAPindel and GATK). Of the 960 indels that were targeted for sequencing, 841 (83.5%) were successfully amplified and sequenced, with an average coverage of 4,866.
Unique-to-GATK indels had a validation rate of 180 of 336 (54.0%), being validated. The validation rate for unique-to-SOAPindel was found to be 44.6%, with 148 of 332 validating. For indels that were called by both SOAPindel and GATK, 132 of 169 (78.1%) were successfully validated (Figure 5).
GATK v2.3-9 and the new HaplotyperCaller
Newer implementations of SNV and indel-calling pipelines continually advance the field of variant discovery and analysis by increasing the accuracy by which variants can be reliably called. Here, we show an example of the differences between previous versions of GATK with respects to SNV calls and indel calls on the same sample, k8101-49685. The vast majority of SNV calls made by both the GATK UnifiedGenotyper version 2.3-9 and the GATK HaplotypeCaller version 2.3-9 modules overlapped with the SNV calls made by the GATK UnifiedGenotyper version 1.5, showing an overall concordance of 91.0% (27,150 of 29,912) and 87.0% (26,751 of 30779) respectively. However, for indel calls, the picture was quite different, with the GATK UnifiedGenotyper version 2.3-9 and GATK HaplotypeCaller version 2.3-9 modules showing an overall concordance with the GATK UnifiedGenotyper version 1.5 calls of 54.7% (1,688 of 3,085) and 54.6% (1,858 of 3,404) respectively (see Additional file 1, Figure S10).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}