
Abstract
Introduction
TNF-like weak inducer of apoptosis (TWEAK) has been proposed as a mediator of inflammation and bone erosion in rheumatoid arthritis (RA). This study aimed to investigate TWEAK and TWEAK receptor (Fn14) expression in synovial tissue from patients with active and inactive rheumatoid arthritis (RA), osteoarthritis (OA) and normal controls and assess soluble (s)TWEAK levels in the synovial fluids from patients with active RA and OA. Effects of sTWEAK on osteoclasts and osteoblasts were investigated in vitro.
Methods
TWEAK and Fn14 expression were detected in synovial tissues by immunohistochemistry (IHC). Selected tissues were dual labelled with antibodies specific for TWEAK and lineage-selective cell surface markers CD68, Tryptase G, CD22 and CD38. TWEAK mRNA expression was examined in human peripheral blood mononuclear cells (PBMC) sorted on the basis of their expression of CD22. sTWEAK was detected in synovial fluid from OA and RA patients by ELISA. The effect of sTWEAK on PBMC and RAW 264.7 osteoclastogenesis was examined. The effect of sTWEAK on cell surface receptor activator of NF Kappa B Ligand (RANKL) expression by human osteoblasts was determined by flow cytometry.
Results
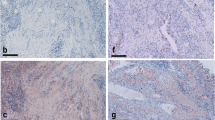
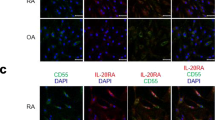
TWEAK and Fn14 expression were significantly higher in synovial tissue from all patient groups compared to the synovial tissue from control subjects (P < 0.05). TWEAK was significantly higher in active compared with inactive RA tissues (P < 0.05). TWEAK expression co-localised with a subset of CD38+ plasma cells and with CD22+ B-lymphocytes in RA tissues. Abundant TWEAK mRNA expression was detected in normal human CD22+ B cells. Higher levels of sTWEAK were observed in synovial fluids isolated from active RA compared with OA patients. sTWEAK did not stimulate osteoclast formation directly from PBMC, however, sTWEAK induced the surface expression of RANKL by human immature, STRO-1+ osteoblasts.
Conclusions
The expression of TWEAK by CD22+ B cells and CD38+ plasma cells in RA synovium represents a novel potential pathogenic pathway. High levels of sTWEAK in active RA synovial fluid and of TWEAK and Fn14 in active RA tissue, together with the effect of TWEAK to induce osteoblastic RANKL expression, is consistent with TWEAK/Fn14 signalling being important in the pathogenesis of inflammation and bone erosion in RA.
Similar content being viewed by others

Introduction
TWEAK (TNF-like weak inducer of apoptosis) is a recently described member of the TNF superfamily. It is reported to exert a variety of biological effects through ligation with its receptor, Fn14. The biological effects of TWEAK include induction of pro-inflammatory cytokines, modulation of the immune response and angiogenesis, stimulation of apoptosis and regulation of tissue repair and regeneration [1, 2]. The pro-inflammatory effects of TWEAK/Fn14 signalling are mediated by several signalling cascades, including NF-B and the mitogen-activated protein kinases (MAPK), ERK1/2, JNK1/2 and p38 [3]. TWEAK induces the production of a large number of pro-inflammatory molecules, such as matrix metalloproteinase (MMP1), IL-6, IL-8, MCP-I and Regulated upon Activation Normal T Cell Expressed and Secreted (RANTES) by synoviocytes and fibroblasts, as well as ICAM-1, E-selectin, IL-8, and MCP-1 by endothelial cells [4]. The majority of these cytokines are induced by TWEAK/Fn14 induction of the NF-κβ signalling pathway [3, 5]. The pro-inflammatory effects of TWEAK are seen in various cell types including glomerular mesangial cells [6], human umbilical vein endothelial cells (HUVEC) [7], human gingival fibroblasts [8], human dermal fibroblasts, synoviocytes [9], chondrocytes, and fibroblasts [2].
Recent reports from us [10] and others [11] are consistent with TWEAK being a key mediator of joint pathology in murine RA models and in human RA [12, 13]. Specifically, recombinant TWEAK enhanced the production of MCP-1 and MIP-2 by synovial cells from collagen induced arthritis (CIA) mice in vitro, while the addition of TWEAK monoclonal antibody ameliorated paw swelling, synovial proliferation and inflammatory cell accumulation in CIA [10, 11]. A role for TWEAK has been described in human RA, where TWEAK induced the proliferation of synovial fibroblasts and increased the production of inflammatory cytokines and chemokines, as well as the expression of ICAM-1 [12]. High serum levels of TWEAK, TNF-α and IL-6 were seen in RA patients as compared to normal controls [13]. Moreover, serum TWEAK levels correlated with the disease activity score (DAS28) in RA patients and high serum TWEAK levels demonstrated a correlation with short-term response to etanercept treatment [13]. Higher levels of TWEAK were found in RA compared to psoriatic synovium [14]. In the current study we examine TWEAK expression in a larger group of patient-derived samples that encompassed active and inactive RA, osteoarthritic (OA) and normal patients. In addition, levels of soluble (s) TWEAK in the synovial fluids of active RA compared with OA patients were determined.
Pertinent to the pathogenesis of cartilage and bone loss in RA, TWEAK has been demonstrated to promote bone and cartilage destruction through inhibition of chondrogenesis, osteogenesis and the induced production of matrix metalloproteinase (MMP)-3 [10, TWEAK expression by PBMC. PBMC from two healthy volunteers were sorted by FACS based on their expression of CD22, yielding CD22+ and CD22- populations of greater than 94% purity based on post-sort analysis (A). Isolated cells were then analysed for TWEAK mRNA expression relative to that of GAPDH, by real-time RT-PCR (B). Data shown are means of triplicate reactions ± SD. Differences in relative expression of TWEAK mRNA between CD22+ and CD22- populations were tested by Student's t-test (**P < 0.001).
TWEAK in synovial fluids from RA and OA
While not statistically significant, there was a trend (P = 0.079) for higher measurable levels of TWEAK protein in active RA synovial fluid (1,226 ± 235 pg/ml) than in the OA samples (713 ± 134 pg/ml) (Figure 5).

Levels of soluble TWEAK in synovial fluids from patients with RA and OA. Scatter plot of sTWEAK levels in the synovial fluids obtained from active RA (n = 17) and OA (n = 16). Horizontal lines reflect mean values of sTWEAK level in each group.
Effect of soluble TWEAK on osteoclastogenesis in vitro
Recombinant human (rh) TWEAK at concentrations as high as 800 ng/ml in the presence of M-CSF did not stimulate osteoclast differentiation from unfractionated PBMC (Figure 6) or from fractionated CD14+ human PBMC (not shown). Furthermore, RANKL/M-CSF osteoclastogenesis was inhibited rather than stimulated by rhTWEAK (Figure 6) as assessed by both TRAP staining and resorption pit formation on dentine slices (Figure 6). Identical results were obtained when murine RAW 264.7 cells were used as osteoclast precursors (data not shown).

Effect of soluble TWEAK on osteoclast formation and function. Human unfractionated PBMC were cultured for nine days in medium containing rhM-CSF only (25 ng/ml) (A), M-CSF and rhTWEAK at 100 ng/ml (B), 400 ng/ml (C) or 800 ng/ml (D), M-CSF and rhRANKL (50 ng/ml) (E), or all of M-CSF (25 ng/ml), RANKL (50 ng/ml) and TWEAK (100 ng/ml) (F). Cultures were then fixed and stained for TRAP. (G) Multinucleated cells (MNC), containing >3 nuclei, positive for TRAP, were counted from quadruplicate wells. Data are expressed as means ± standard deviation. Significant differences were determined by one way analysis of variance (ANOVA) with Tukey post-hoc test: a indicates difference to M-CSF only control (P < 0.001) and b denotes difference to M-CSF+RANKL treatment (P < 0.05). (H) PBMC were seeded onto dentine slices in the presence of rhM-CSF (25 ng/ml) with the addition of rhRANKL (50 ng/ml) and/or rhTWEAK as indicated. Resorption was assessed after 14 days of culture by SEM and is expressed as the mean ± SD resorption expressed as a percentage of that measured for the RANKL/M-CSF control. Data shown are pooled from two independent experiments with resorption assessed for four dentine slices/treatment/donor. No significant differences were observed between RANKL treatments.
Effects of soluble TWEAK on cell surface RANKL expression
Treatment of human primary osteoblasts with rhTWEAK for three days resulted in the increased cell surface expression of RANKL. Furthermore, RANKL expression was induced on a population of osteoblasts expressing the immature osteoblast marker, STRO-1 (Figure 7). Together, these findings are consistent with the induction by TWEAK of a pro-osteoclastogenic osteoblastic phenotype.

Effect of soluble TWEAK on cell surface RANKL expression by STRO-1 osteoblast subpopulation. Human primary osteoblasts were cultured untreated or treated with rhTWEAK (50 ng/ml) for three days, harvested by collagenase/dispase digestion and stained for STRO-1 and RANKL expression. Stained cells were analysed by FACS. The percentage of cells present in each of the four sub-populations is indicated. Data are presented for two independent donors' cells.
Discussion
The findings of this study are consistent with the growing evidence that TWEAK is a mediator of the joint destruction both in animal models of RA [10, 11] and in human RA [12]. Recently, a number of cell types involved in the pathogenesis of RA have been reported to express TWEAK and its receptor, Fn14. While synovial fibroblasts from human patients with active RA have been reported to express TWEAK and Fn14 [14] another report demonstrated TWEAK expression by an unidentified CD45-positive haemopoietic cell population and Fn14 expression by both CD45-positive and -negative cells [12]. In contrast, van Kuijk and co-workers [14] reported TWEAK/Fn14 expression by macrophages but not by lymphocytes. The current study demonstrates that several cell types in RA synovial tissue express TWEAK, including CD68-positive macrophages, confirming the results of van Kuijk and co-workers [14]. Interestingly and in contrast to van Kuijk and co-workers, we observed that a sub-population of CD22-positive B lymphocytes and CD-38 positive plasma cells found in lymphocyte aggregates in the sub-lining synovial tissues expressed TWEAK. Our findings are consistent with those of Kraan and coworkers who found that the numbers of CD38+ plasma cells and CD22+ B cells in RA were the best discriminating markers when comparing RA to non-RA inflammatory synovial samples [22].
In the current study, TWEAK expression by plasma cells is in agreement with our recent finding that demonstrated the expression of TWEAK in bone marrow plasma cells in patients with multiple myeloma [18]. In that study, serum levels of TWEAK correlated with the serum bone resorption marker, β-crosslaps, and levels of the osteoclast recruitment chemokine, CXCL12, strongly implying a pathological role for plasma cell-derived TWEAK in bone erosion associated with multiple myeloma [18]. In the current study, we found that the TWEAK-positive B-Lymphocytes in the germinal centres of normal tonsil sections were rare, consistent with the infrequency of CD22+ B cells at this site [29]. While the CD22+TWEAK+ expressing B cells may be a specific population associated with the chronic inflammation seen in RA, CD22-positive B lymphocytes isolated from human normal peripheral blood also expressed abundant levels of TWEAK mRNA, suggesting that B-lineage cells are a source of TWEAK. Based on our findings in multiple myeloma [18] these cells may possibly also be associated with the bone erosion characteristic of RA. Interestingly, an immunotherapeutic approach that targets CD22, epratuzumab, has proved efficacious in the treatment of non-Hodgkin lymphoma, and in the autoimmune diseases, systemic lupus erythematosus and primary Sjögren's syndrome [30]. Our findings suggest that CD22 and CD38 are also potential therapeutic targets in RA.
We were unable to verify the cell types expressing Fn14 using dual label IHC for technical reasons; however, many of these cells also had the morphology of lymphocytes and monocytes and importantly our observation indicates that Fn14 was strongly expressed by multinucleated cells within the synovial tissue. We have previously demonstrated that these multinucleated cells were preosteoclasts [31]. It is well known that the active rheumatic joint contains an influx of osteoclast precursors [31]. This is also consistent with a recent study by our group demonstrating the expression of TWEAK and Fn14 by multinucleated cells in gingival tissues from chronic periodontitis patients [32]. This would suggest that TWEAK-Fn14 signalling may stimulate osteoclastogenesis and hence could contribute to the bone loss associated with chronic inflammatory diseases [10, 11].
The observation that Fn14 is expressed by cells associated with blood vessels is consistent with the report that TWEAK regulates human endothelial cell proliferation, migration and survival in vitro [33, 34]. Both TWEAK and Fn14 expression was associated with synovial tissue blood vessels suggesting that TWEAK has an autocrine effect on the angiogenesis seen in RA synovial tissue. Furthermore, this is consistent with the perivascular expression of TWEAK and Fn14 expression in RA and psoriatic synovial tissue [14].
A significant decrease in serum TWEAK has been reported in RA patients responding to treatment with etanercept [13, 14] while persistent TWEAK/Fn14 expression was seen in a RA patient cohort receiving infliximab treatment [14] suggesting a role for TWEAK in modulation of disease activity. Our data demonstrated significantly lower expression of TWEAK in the inactive RA synovial tissues. In the majority of patients this followed successful non-TNF targeted DMARD treatment, suggesting that DMARDs may reduce TWEAK expression as seen in the etanercept-treated patients. Higher TWEAK expression in active RA was also reflected in the increased synovial fluid levels of sTWEAK compared with those in OA in our study. Based on this, we suggest that monitoring the sTWEAK levels in synovial fluids could possibly be another marker of disease progression.
It was previously reported that TWEAK stimulates the differentiation of RAW 264.7 monocyte/macrophage cells into functional osteoclasts, and did so in the absence of Fn14 expression [17]. However, we could not reproduce these findings in vitro using recombinant TWEAK to directly stimulate osteoclast formation from RAW 264.7 cells, or from human PBMC, either unfractionated or sorted on the basis of CD14 expression, a fraction shown previously to contain the osteoclast precursor [25, 35]. Although the present study demonstrated that multinucleated cells expressed Fn14, our data indicate that the stimulation of osteoclast formation by TWEAK may not be due to a direct interaction with osteoclast precursors. However, we have demonstrated that stromal osteoblasts express Fn14 and are sensitive to the effects of TWEAK [10, 16], including the induced expression of RANKL and CXCL12 mRNAs [18]. This supports an indirect role for TWEAK in recruiting and generating osteoclasts, via the osteoblast. In the current study we confirmed that TWEAK induced the expression of cell surface RANKL protein and did so in a population of human osteoblasts expressing the immature osteoblast cell surface marker, STRO-1, cells we have previously demonstrated are capable of supporting osteoclastogenesis [36]. This is reminiscent of our earlier study, in which RANKL mRNA expression was preferentially induced in STRO-1-positive osteoblasts in response to 1α,25-dihydroxyvitaminD3 [37]. In addition, we recently reported a novel role for TWEAK in the regulation of osteoblast differentiation [16] and showed that TWEAK, and the combination of TWEAK and TNFα, induced the expression of sclerostin, a negative regulator of bone formation [38], and as we recently demonstrated [39], human osteoblast differentiation and matrix mineralization. Thus, the elevated TWEAK expression we see in RA may not only stimulate bone resorption by osteoclasts by induction of RANKL on osteoblasts but also inhibit new bone formation, contributing to the reduction in bone mass observed in inflammatory bone disease [40].
Conclusions
The current study found high levels of TWEAK expression in synovial tissues and synovial fluids from patients with active RA. Together with its ability to induce cell surface expression of RANKL by osteoblasts and possibly induce activation of B cells into plasma cells we postulate that TWEAK in this context would be pro-osteoclastogenic and would thus contribute to the bone loss associated with this chronic inflammatory disease. These findings support the concept that TWEAK should be considered as a therapeutic target in RA.
{kind=link}