
Abstract
Background
The filamentous fungus Trichoderma reesei is an important host organism for industrial enzyme production. It is adapted to nutrient poor environments where it is capable of producing large amounts of hydrolytic enzymes. In its natural environment T. reesei is expected to benefit from high energy yield from utilization of respirative metabolic pathway. However, T. reesei lacks metabolic pathway reconstructions and the utilization of the respirative pathway has not been investigated on the level of in vivo fluxes.
Results
The biosynthetic pathways of amino acids in T. reesei supported by genome-level evidence were reconstructed with computational carbon path analysis. The pathway reconstructions were a prerequisite for analysis of in vivo fluxes. The distribution of in vivo fluxes in both wild type strain and cre1, a key regulator of carbon catabolite repression, deletion strain were quantitatively studied by performing 13C-labeling on both repressive carbon source glucose and non-repressive carbon source sorbitol. In addition, the 13C-labeling on sorbitol was performed both in the presence and absence of sophorose that induces the expression of cellulase genes. Carbon path analyses and the 13C-labeling patterns of proteinogenic amino acids indicated high similarity between biosynthetic pathways of amino acids in T. reesei and yeast Saccharomyces cerevisiae. In contrast to S. cerevisiae, however, mitochondrial rather than cytosolic biosynthesis of Asp was observed under all studied conditions. The relative anaplerotic flux to the TCA cycle was low and thus characteristic to respiratory metabolism in both strains and independent of the carbon source. Only minor differences were observed in the flux distributions of the wild type and cre1 deletion strain. Furthermore, the induction of the hydrolytic gene expression did not show altered flux distributions and did not affect the relative amino acid requirements or relative anabolic and respirative activities of the TCA cycle.
Conclusion
High similarity between the biosynthetic pathways of amino acids in T. reesei and yeast S. cerevisiae was concluded. In vivo flux distributions confirmed that T. reesei uses primarily the respirative pathway also when growing on the repressive carbon source glucose in contrast to Saccharomyces cerevisiae, which substantially diminishes the respirative pathway flux under glucose repression.
Similar content being viewed by others


Background
The industrially important protein producer, the filamentous fungus Trichoderma reesei, a clonal derivative of the ascomycete Hypocrea jecorina, is adapted to growth in nutrient poor environments, where it is able to use complex plant material as carbon source. T. reesei and a number of other filamentous fungi and cellulolytic bacteria produce and secrete plant polymer hydrolyzing enzymes, such as cellulases and hemicellulases, into their surroundings to break down the polymers into easily metabolizable monomers [1].
Because of its ability to synthesize and secrete large amounts of proteins, T. reesei has gained industrial importance in production of enzymes of native and heterologous origin. Carbon catabolite repression (CCR) of T. reesei negatively regulates the powerful production machinery of the hydrolytic enzymes when a preferred carbon source, such as glucose, is available. Inducers of hydrolytic enzyme expression are often small oligosaccharides or derivative parts of the polymers from the environment of the fungus. The inductive signaling leads to synthesis of specific sets of enzymes [2, 3]. In T. reesei, D-xylose, xylobiose, sophorose, and lactose have been observed to trigger production of particular enzyme sets [4, 5]. Sophorose, a molecule of two beta-1,2-linked glucose units, is an efficient inducer of cellulose gene expression at low concentration (1-2 mM) when T. reesei is growing on a non-repressing carbon source, such as sorbitol or glycerol [6]. However, in high glucose concentrations CCR overrules the inductive signals in T. reesei [6].
Sorbitol as a carbon source neither provokes CCR nor triggers the cellulase gene expression in T. reesei [6]. Nevertheless, cellulase production is positively correlated with the ability of different T. reesei strains to grow on D-sorbitol [7], which could be converted to L-sorbose [8] that induces cellulase expression in T. reesei [9]. In T. reesei L-arabinitol 4-dehydrogenase (Lad1) is involved in the initial oxidization of D-sorbitol at C2 to convert it to D-fructose [10]. A specific sorbitol dehydrogenase converts sorbitol to fructose in Aspergilli fungi [11, 2].
Cre1 is the key mediator protein of CCR in T. reesei [12, 13]. Trichoderma Cre1 has a 95% sequence similarity with Aspergillus CreA in regions of the zinc-finger and proline-serine-threonine-rich domain and the complete sequences are 46% identical [13]. Cre1 is structurally also highly similar to Mig1, a key protein in glucose repression in yeast Saccharomyces cerevisiae [12, 13]. However, the functional dissimilarities observed between Cre1/CreA and Mig1, in spite of the sequence and structural similarity, have led to the conclusion that glucose repression functionalities in filamentous fungi and yeasts have evolved separately [14, 15]. Pfeiffer et al argued that the evolution from unicellular to undifferentiated multicellular organisms, like T. reesei, has been facilitated by the general preference of high yield energy generation through respiration even in the presence of a preferred carbon source [16]. In contrast to CCR regulation in S. cerevisiae, it has been shown that in T. reesei CCR does not cause repression of genes encoding the TCA cycle enzymes or respiratory pathway components [17, 18]. David et al observed differences in the distribution of intracellular carbon fluxes in central carbon metabolism between A. nidulans reference and a carbon repression deletion mutant (creA Δ4) strains when they were grown on glucose [19].
Despite the industrial importance of T. reesei, its genome has only recently been sequenced [ Origins of proteinogenic amino acids. The origins of the carbon backbones of the proteinogenic amino acids utilized in METAFoR analysis [26] and for which the biosynthetic pathways were reconstructed by computational pathway analysis method ReTrace [21]. The amino acids for which the biosynthetic pathway was not directly found by ReTrace are denoted in red italics. The amino acid carbons are denoted in the following way: a = α, b = β, g = γ, d = δ, e = ε, ksi = ξ.
The fungal biosynthetic pathway of Lys from Oga [29] was not found by ReTrace because of inconsistencies in the atom map** in the KEGG reaction database. However, because the reactions of the alternative biosynthetic route of Lys which is active, for example in bacteria, did not gain good scores for presence in T. reesei, the fungal pathway was assumed prior to the 13C-pathway analysis. Pathways from Oga to Arg were identified by ReTrace but the pathway known to be active in S. cerevisiae was not found among them. Most of the identified pathways were directed through 1-pyrroline-5-carboxylate dehydrogenase reaction (1.5.1.12) in the reverse direction, which forms a false shortcut path between Oga and Arg.
After the unsuccessful direct search of pathway from Oaa to Thr, Thr biosynthesis pathway was searched from Asp, an intermediate in the pathway from Oaa to Thr. Genome level evidence of the presence of the pathway was found. The biosynthetic pathway of Ile that is active in S. cerevisiae was not found directly from precursors Oaa and Pyr because the pathway proceeds first from Oaa to Thr and that pathway was not directly identified as discussed above. The reactions further from Thr were identified with high scores for genome level evidence of their presence in T. reesei and thus, the pathway that is known to be active in S. cerevisiae is evidently present also in T. reesei. Tyr biosynthesis pathway was found from precursors downstream to 3-(4-hydroxyphenyl)pyruvate and only the transamination was lacking from a complete pathway. However, a high scoring hit for a transaminase sequence was separately searched and identified in the genome of T. reesei. Most of the high scoring alternative pathways could be excluded because only the anabolic pathways are active in the exponential growth and in absence of amino acids in the medium.
13C-pathway analysis and prediction of subcellular localization of key enzymes
The pathways of amino acid biosynthesis reconstructed in T. reesei corresponded to the pathways utilized by S. cerevisiae. The fragmentomer data from 13C-labeling of proteinogenic amino acids provided further confirmation for this (see Methods for the definition of fragmentomer data). The 13C-labeling patterns of the carbon backbones of proteinogenic amino acids originate from the 13C-labeling of their precursor metabolites in central carbon metabolism and thus, the 13C-labeling patterns of amino acids can be propagated to the precursors to identify the active pathways. In particular, the Lys 13C-labeling pattern indicated its synthesis from Oga via α-aminoadipate pathway, as in yeasts [30]. However, in contrast to S. cerevisiae and a number of other yeast [26, 31], the 13C-labeling pattern of Asp indicated that it primarily originated from mitochondrial Oaa under all the studied conditions (Figure 2). Mitochondrial Asp synthesis has previously been observed in Yarrowia lipolytica [31]. Furthermore, identical 13C-labeling patterns were observed in Asp and Thr. This confirmed Thr synthesis from Asp and excluded a contribution from the reversible threonine aldolase reaction [32].

Comparison of the fractions of corresponding intact bonds in amino acid precursors Oaa, Oga and Pep in T. reesei. The data is taken from all replicates of fractional [U-13C]glucose or [U-13C]sorbitol experiments performed (1-3 wild type on glucose, 4-6 Δcre1 on glucose, 7-9 wild type on sorbitol, 10-12 wild type on sorbitol (sophorose experiment control), 13-15 wild type on sorbitol (sophorose induction), 16-17 Δcre1 on sorbitol (sophorose experiment control), 18-19 Δcre1 on sorbitol (sophorose induction)). Oaa data was detected from Asp-Cα, -Cβ and Thr-Cα, Oga data from Glu-Cα, -Cβ and Pro-Cα, -Cβ and Pep data from Phe and Tyr-Cα. Fractions of intact bonds in Oaa, Oga and Pep were calculated from combinations of fragmentomers. A) OAA_x1x is the fraction of Oaa molecules with an intact bond at C2-C3, OGA_x1xx is the fraction of Oga molecules with an intact bond at C2-C3 and PEP_x1 is the fraction of Pep molecules with an intact bond at C2-C3. B) OAA_xx1 is the fraction of Oaa molecules with an intact bond at C3-C4 and OGA_1xxx is the fraction of Oga molecules with an intact bond at C1-C2. Error bars are ± SEMs. The carbon chain of Oaamit remains intact in the TCA cycle except that C1 is cleaved in the synthesis of Oga. Almost the entire labeling pattern of Oaamit can be assessed from the labeling pattern determined for Oga. If Asp and Thr synthesis originates from Oaamit, the fractions of corresponding Oaa and Oga intact fragments in the figures should match.
A three carbon fragment of mitochondrial Oaa (Oaamit) (C2-C3-C4) remains intact in the synthesis of the TCA cycle intermediate Oga and therefore the 13C-labeling pattern of Oaamit can be partially observed in Glu that originates from Oga. In the exponential growth phase it is reasonable to assume unidirectional transport of Oaa across the mitochondrial membrane, which has previously been experimentally shown in S. cerevisiae [26]. When the backward transport from Oaamit to Oaacyt is negligible, a three-carbon fragment of Oaacyt (C1-C2-C3) is produced from Pep, a precursor of Phe and Tyr, via glycolysis and by pyruvate carboxylase. The fractions of intact Cα-Cβ bonds in Asp and Thr were highly similar to the corresponding intact carbon fragments in Oga, propagated from Glu, but clearly different from the corresponding intact carbon fragments in Pep, propagated from Phe, Tyr in T. reesei, indicating the primarily mitochondrial origin of Asp. Since the C3-C4 bond of Oaamit remains intact in the TCA cycle, Oaamit serving as a precursor for Asp and Thr biosynthesis was further supported by the high similarity in the fraction of molecules having the corresponding C-C fragment intact, i.e. the C3-C4 fragment of Oaa, propagated from Asp, Thr, and C1-C2 fragment of Oga, propagated from Glu, (OAA_xx1 and OGA_1xxx, respectively, Figure 2).
Additional support for mitochondrial Asp synthesis was obtained from sequence analysis. Evidence of mitochondrial targeting peptide sequence was identified in one of the T. reesei genome sequences with homology to the aspartate aminotransferases in S. cerevisiae by TargetP analysis [33, 34] (Additional file 2). This strongly supported the mitochondrial localization of one of the encoded enzymes. However, no evidence of mitochondrial targeting peptide was identified by TargetP analysis of the T. reesei sequence with homology to the S. cerevisiae pyruvate carboxylase that produces Oaa. Thus, Oaamit could originate both from transport across the mitochondrial membrane and from the TCA cycle.
Pyruvate is a precursor of Ala and Val biosynthesis. If the pyruvate pools in cytosol and mitochondria possess significantly different 13C-labeling patterns, for example as a result of malic enzyme flux, a mitochondrial localization of pyruvate-based amino acid synthesis can be confirmed from the 13C-labeling data [31]. However, the fractions of intact two-carbon fragments Pyr C1-C2 and C2-C3 observed in Ala and Val and the corresponding two-carbon fragments in Pep, a direct precursor of Pyrcyt, observed in Phe and Tyr, were not significantly different under the studied conditions. Therefore, the 13C-labeling patterns could not be utilized to assess the localization of the synthesis of pyruvate-based amino acids. Strong evidence of a mitochondrial targeting sequence in the T. reesei sequence that showed homology to the acetolactate synthase in S. cerevisiae was identified by TargetP [33, 34] (Additional file 2). In yeast the first enzyme in Val biosynthesis, the acetolactate synthase, has been reported to be localized in mitochondria [35], whereas cytosolic and mitochondrial isoenzymes of alanine aminotransferase have been observed [30].
Ser originates from glycolytic intermediate 3-phosphoglycerate and can be further converted to Gly and a C1 unit by the reversible reaction of serine hydroxymethyl transferase (SHMT). Gly could also originate from threonine aldolase or from the reversible reaction of the glycine cleavage pathway (i.e., C1 + CO2). In S. cerevisiae glycine cleavage pathway is active inside mitochondria [36] and although both mitochondrial and cytosolic isoenzymes of SHMT exist in S. cerevisiae [37, 38], the effect of the glycine cleavage pathway on the Ser-Cα f(1) fraction has not been observed in S. cerevisiae batch cultures grown on glucose [26]. In T. reesei the activity of the glycine cleavage pathway was observed in the 13C-labeling pattern of Ser, since Ser-Cα f(1) fragmentomer fraction of Ser molecules with both carbon bonds cleaved was higher than the fraction of fully cleaved Pep, a three carbon lower glycolytic intermediate. The fraction of fully cleaved Pep molecules was observed in Phe and Tyr-Cα f(1) fragmentomer fractions (Figure 3). Two T. reesei sequences were observed to have homology to the S. cerevisiae SHMT sequences. In one of them a strong evidence of a mitochondrial targeting pre-sequence was found by TargetP [33, 34] (Additional file 2). Therefore, SHMT activity likely occurs in both cytosolic and mitochondrial compartments of T. reesei. The Ser 13C-labeling pattern observed in T. reesei indicates either a partially cytosolic localization of the glycine cleavage pathway or protein synthesis occurring primarily from a mitochondrial pool of Ser. TargetP analysis of the T. reesei sequence homological to sequence of the S. cerevisiae glycine dehydrogenase, the p-subunit of the Gly cleavage system, showed no clear indication of a mitochondrial targeting pre-sequence [33, 34] (Additional file 2).

Effect of the reversible glycine cleavage pathway. Effect of the reversible glycine cleavage pathway in T. reesei wild type (wt) and Δcre1 strains on the 13C-labeling pattern of Ser. The fraction of Ser-Cα f(1) fragmentomer from the total pool of Ser, compared to the corresponding fraction in Pep (C2) observed from Phe and Tyr-Cα f(1) fragmentomers. Error bars are ± SEMs.
Amino acids belonging to the Glu amino acid family, Glu, Pro and Arg, showed a highly similar 13C-labeling, as expected, in both strains grown on glucose and in the wild type strain culture grown on sorbitol (data not shown). In contrast, a significant variation was observed in the Glu, Pro and Arg fragmentomers in cultures when their pre-cultures were mixed before sophorose induction experiment. This observation may be explained by differential mobilization of amino acids from cellular compartments resulting from the perturbation when the cultures were mixed prior to the induction period.
METAFoR analysis of T. reesei
The relative fluxes merging at the branching points of central carbon metabolism of T. reesei were determined by BDF 13C-labeling with glucose and sugar alcohol sorbitol as carbon sources. The flux ratios of the wild type and Δcre1 T. reesei strains determined in batch cultures on glucose, on sorbitol and on sorbitol with sophorose induction of expression of cellulase genes are shown in Table 2.
Respiratory pathway flux of T. reesei
The relative anaplerotic flux (the fraction of OAAmit from Pep, Table 2) describes the relative activities of the biosynthetic and respirative carbon fluxes in the TCA cycle. The anaplerotic flux replenishes carbons to the TCA cycle flux by importing C4 compounds as there is drain of carbon to biosynthesis. David et al concluded that the TCA cycle was more active in ΔcreA mutants of A. nidulans than in the wild type when grown on glucose [19]. However, they suggested that it resulted from a higher ATP demand in the deletion strain possibly caused by active futile cycles instead of derepression of the respirative pathway flux. No significant difference was observed in the anaplerotic flux ratios between the T. reesei wild type and Δcre1 strains grown on glucose. Thus, in T. reesei the TCA cycle was as active relative to biosynthesis in the Δcre1 strain as in the wild type strain and so, Cre1 does not mediate repression of respirative pathway flux in T. reesei either.
A difference was observed between the two T. reesei strains when grown on sorbitol. The relative anaplerotic flux was 26% in the wild type strain and 42% in the Δcre1 strain (Table 2). This may indicate that there was a difference in the specific growth rates of the two strains on sorbitol.
Previously, an excess of glucose has been found to only partially repress the gene expression of the enzymes of the TCA cycle and the components of the respiratory chain in T. reesei [17]. That is in contrast to the effect of excess of glucose on S. cerevisiae, where glucose repression extensively downregulates the respiratory pathway at the transcriptional level [18]. The anaplerotic flux ratio in T. reesei wild type strain was higher on glucose (35%) than that on sorbitol (26%) (Table 2). The results indicated a higher activity of respiratory metabolism relative to biosynthesis on the non-repressing carbon source sorbitol than that on the repressing carbon source glucose. A complete oxidation of sorbitol, that is a more reduced carbon source than glucose, results in a higher relative flux of electrons per carbon source molecule to the respiratory chain than during growth on glucose. Thus, if T. reesei respired at maximum rate during the batch growth on glucose, fluxes producing reduced cofactors, for example biosynthetic pathway fluxes or the TCA cycle fluxes, would have decreased on sorbitol.
Small fractions of Pyrmit originating from malate via the action of the malic enzyme were observed in both strains under almost all conditions (Table 2).
Pentose phosphate pathway (PPP) of T. reesei
A lower fraction of triose phosphates originated from pentose phosphates in the wild type strain (39%) than in the Δcre1 strain (47%) when grown on glucose (Table 2). In batch cultures under excess glucose conditions, the gluconeogenesis by phosphoenolpyruvate carboxykinase and the reverse transport of Oaa across the mitochondrial membrane are assumed to have negligible fluxes [18, 26]. For this purpose the fraction of Pep originating from the pentose phosphate pathway (PPP) was calculated neglecting any contribution of phosphoenolpyruvate carboxykinase to the 13C-labeling pattern of Pep, propagated from Phe, Tyr. The fraction of Pep originating from PPP represents the flux via PPP relative to the total flux to Pep. However, this fraction is not a direct measure of the flux through the oxidative branch of the PPP but includes molecules that have only gone through reversible reactions in the non-oxidative PPP. Furthermore, the standard deviation is always high because only 40% of the triose phosphates that originate from the PPP have different 13C-labeling patterns than the triose phosphates originating from glycolysis.
The differences in the relative flux through the PPP to the triose phosphates can be caused by differences in the glycolytic rate or in NADPH demands, since the oxidative branch of the PPP is usually the main source of cytosolic NADPH. A low glycolytic rate could allow the label scrambling in the non-oxidative part of the PPP to affect the 13C-labeling patterns of a large fraction of triose phosphates.
The reversible fluxes through the reactions of transketolase and transaldolase, observed in the 13C-labeling patterns of pentose phosphates that can be detected in His, were clearly different in glucose and sorbitol cultivations (Table 2). The fraction of pentose phosphates that had gone through a transketolase reaction (R5P from T3P and S7P) was 51% and 42% when glucose was the carbon source for the wild type and the Δcre1 strains, respectively. When grown on sorbitol the fractions were higher, 72% and 79% for the wild type and the Δcre1 strains, respectively. The fraction of pentose phosphates cleaved in the transaldolase and transketolase reactions (R5P from E4P) was 25% and 23% when grown on glucose, whereas when sorbitol was the carbon source they were 46% and 54% for the wild type and the Δcre1 strains, respectively. The higher fractions of pentose phosphates cleaved in the reactions of transketolase or transaldolase when grown on sorbitol could be a result of entrance of sorbitol in the central carbon metabolism and into the PPP directly in a form of fructose 6-phosphate [10].
Figure 4 shows the relative abundances of the contiguous 13C-fragments around His-Cβ, which originate from fragments around ribose 5-phosphate C3. When sorbitol was the carbon source lower fractions of fully intact His fragments and higher fractions of His fragments cleaved in the reversible reactions of transaldolase and transketolase were observed in both strains than when grown on glucose. This indicated higher relative fluxes in the non-oxidative part of the PPP when compared to the rate of withdrawal of pentose phosphates to His biosynthesis. When sorbitol was the carbon source the relative activity of the non-oxidative PPP compared to the rate of biosynthetic drain of pentose phosphates was even higher in the Δcre1 strain than in the wild type strain. The fraction of fully cleaved His-Cβ f(1) fragments was higher in the Δcre1 strain than in the wild type strain when they were grown on sorbitol (Figure 4). Correspondingly, lower fractions of fully intact His-Cβ f(3) were observed in the Δcre1 strain than in the wild type strain.

13C-His-Cβ centered contiguous 13C-fragments in T. reesei. Fractions of 13C-His-Cβ centered contiguous 13C-fragments in T. reesei in different genetic, wild type (wt) and Δcre1 mutant. Cultures were grown on glucose (glucose repressed) or sorbitol (derepressed). Sorbitol grown cultures were grown with or without induction of cellulase gene expression by the addition of sophorose to some cultures. His-Cβ f(1) denotes fragments with C-C bonds cleaved on both sides of Cβ, His-Cβ f(2) and f(2*) denote fragments with Cβ-Cα and Cβ-Cγ preserved, respectively, and His- Cβ f(3) denotes fragments were both bonds are intact. Error bars represent ± SEM.
Effect of sophorose induction of cellulase gene expression on metabolic fluxes
Induction of cellulase gene expression with sophorose did not cause any significant changes in the metabolic flux distributions in the central carbon metabolism of T. reesei. Therefore, the induction of cellulase gene expression did not affect the relative fluxes to different amino acid families or the ratio of anabolic and catabolic activity of the central carbon metabolism. Alteration in the relative biosynthetic rates of different amino acids would have occurred if the amino acid composition of the induced cellulases had been significantly different from the amino acid composition of the proteins generally produced by T. reesei which was not observed.
Flux ratio profiles of T. reesei, S. cerevisiae and Pichia stipitis indicate differences in preferred utilization of pathways
The anaplerotic flux ratios determined hereby in the wild type and Δcre1 T. reesei strains in batch cultures, both grown on glucose, were substantially lower and similar to what has been observed in fully respiratory metabolism in S. cerevisiae in glucose-limited chemostat cultures, where there is no glucose repression [27] (Table 3). The extensive glucose repression of the TCA cycle and the respiratory pathway activity in S. cerevisiae result in high anaplerotic ratio in batch cultures on glucose [26]. The anaplerotic flux ratios in the T. reesei strains with glucose as a carbon source were also similar to the ones observed in P. stipitis, both when grown on glucose in batch cultures and in glucose-limited chemostat cultures [27]. P. stipitis completely lacks aerobic alcoholic fermentation.
It has previously been determined that glucose does not cause extensive repression of the gene expression of the TCA cycle and the respiratory pathway components in T. reesei [17] as it does in S. cerevisiae [18]. The 13C-labeling and METAFoR analysis results on the level of in vivo fluxes confirmed that for highly efficient energy generation through complete oxidation of carbon source T. reesei indeed uses primarily the respirative pathway also when growing on a preferred carbon source glucose. The regulatory differences between T. reesei and S. cerevisiae have been explained as adaptation to different growth environments. S. cerevisiae is adapted to nutrient rich environments in which it has competitive advantage from fast nutrient utilization and a high rate of ATP production through the fermentative pathway, whereas T. reesei is adapted to nutrient poor environments where it benefits from high energy yield [17, 16]. It has also been postulated that undifferentiated multicellular organisms, of which T. reesei is an example, have gotten evolutionary advantage from preferring the high energy yield from respiratory metabolism [16].
Conclusion
Biosynthetic pathways of T. reesei were reconstructed for most of the proteinogenic amino acids by using a computational carbon path analysis method ReTrace. The method was used to search for pathways from a metabolic network consisting of all reactions found in a comprehensive metabolic reaction database, and to subsequently rank the pathways according to the degree of support from the T. reesei's genome [21]. Contiguous pathways, identical to the amino acid biosynthetic routes of S. cerevisiae, were found with high genome-level evidence. The 13C-labeling patterns observed in this study were in good accordance with the compartmentalized model of eukaryotic central carbon metabolism, originally developed for S. cerevisiae [26]. However, in contrast to S. cerevisiae, Asp synthesis was observed to occur primarily from the mitochondrial pool of Oaa in both T. reesei strains under all the studied conditions.
The T. reesei wild type strain is known to exhibit carbon catabolite repression of hydrolytic gene expression during growth on glucose, whereas in the Δcre1 strain the repression is partially disturbed [13]. The respirative pathway in T. reesei does not become transcriptionally downregulated by the carbon catabolite repression as in S. cerevisiae [17]. However, it is the in vivo fluxes that are the ultimate phenotype of an organism. In the present work, the effect of carbon catabolite repression on in vivo fluxes in T. reesei was, for the first time, quantitatively studied. The relative anaplerotic flux to the respirative pathway flux was characteristic to primarily respiratory metabolism in the both T. reesei strains under all studied conditions. Thus, T. reesei utilizes primarily respiratory metabolism also when growing on a preferred carbon source glucose. However, the observed relative anaplerotic fluxes suggested that the respirative activity of the TCA cycle is even slightly higher when T. reesei grows on the neutral carbon source sorbitol than when it grows on glucose. Only minor differences were observed between the in vivo flux distributions of the wild type and the Δcre1 T. reesei strains. This indicates, that Cre1, the key repressor of utilization of alternative carbon sources, does not mediate carbon source dependent metabolic state alterations in the central carbon metabolism of T. reesei. The induction of cellulase gene expression with sophorose did not result in significant changes in the relative requirements of proteinogenic amino acids or in the ratio of anabolic and oxidative activities of the TCA cycle.
Methods
Strains, media and culture conditions
Biosynthetically directed fractional (BDF) 13C-labeling of proteins was carried out for the T. reesei QM6a (wild type) [39] and T. reesei QM6a with deleted cre1 gene (unpublished). Both strains were cultivated in triplicate on two different carbon sources: glucose and sorbitol. Glucose cultivations were carried out in 2 l flasks in 200 ml of minimal medium ((NH4)2SO4 7.6 g/l, KH2PO4 15.0 g/l, 2.4 mM MgSO4, 4.1 mM CaCl2, CoCl2 3.7 mg/l, FeSO4·7H2O 5 mg/l, ZnSO4·7H2O 1.4 mg/l, MnSO4·7H2O 1.6 mg/l, pH adjusted to 4.8 with KOH) supplemented with 2% (w/v) glucose containing 10% (w/w) [U-13C]glucose.
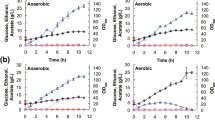
The 200 ml cultures were inoculated with 8 × 107 spores and cultivated at +28°C with constant agitation at 250 rpm. After 35 h of cultivation, during the exponential growth phase (Figure 5), 30 ml and 50 ml samples were withdrawn for dry weight determination and for NMR experiments, respectively. Mycelium from the samples was collected by filtration through Whatmann GF/B filters and washed twice with the sampling volume of water. For dry weight determination the mycelium was dried in an oven at +106°C overnight and weighed.

T. reesei growth curves. Growth curves of T. reesei wild type (wt) and Δcre1 strains (A) on glucose and (B) on sorbitol. Error bars are standard deviations of three replicates.
The BDF 13C-labeling of the wild type strain on sorbitol was carried out in three replicates with 2% (w/v) sorbitol containing10% (w/w) [U-13C]sorbitol, similarly as in the glucose cultivations. After 104 h of incubation, in the exponential growth phase (Figure 5), 30 ml and 50 ml samples were withdrawn for dry weight determination and for NMR experiments, respectively.
BDF 13C-labeling on sorbitol was also carried out with induction of cellulase gene expression by sophorose. Six 2 l flasks of each strain were inoculated, with 2% (w/v) sorbitol as the sole carbon source in minimal medium (see above). After 76 h for the wild type and after 114 h for the Δcre1 mutant, in exponential growth phase (Figure 5), the six cultures were combined, a 30 ml sample for dry weight determination was withdrawn and then the culture broth was redivided into six flasks. The final concentration of 1 mM sophorose was introduced into three of the six replicate 2 l flasks to induce cellulase gene expression. An identical volume of water was added to the three control cultures. Three hours after the induction, when cellulase gene expression was expected to be at a moderate level [6], 0.4 g of [U-13C]sorbitol was added to all six cultures to initiate BDF 13C-labeling. The addition of 0.4 g of [U-13C]sorbitol at this time was estimated to result in a [U-13C]sorbitol fraction of about 10% of the total sorbitol in the culture medium. After 24 h from the addition of the [U-13C]sorbitol, still during the early-exponential growth phase (Figure 5), 30 ml and 50 ml samples were withdrawn for dry weight determination and NMR experiments, respectively. Thereby the 13C-labeled fraction of biomass was synthesized in the induced conditions and the information of the pathways that were active when the cellulase gene expression was induced was recorded in the labelling patterns of proteinogenic amino acids.
Nuclear Magnetic Resonance (NMR) spectroscopy experiments
The filtered mycelial samples were suspended into 10 ml of 6 M HCl and the biomass was hydrolysed in sealed glass tubes at +110°C for 22 h. The suspensions were dried and dissolved in H2O for filtration through 0.2 μm filters. The filtrates were vacuum-dried and dissolved in D2O for NMR experiments. The pH of the samples was below 1 due to residual HCl.
13C-HSQC NMR spectra were acquired at +40°C on a Varian Inova spectrometer operating at a 1H-resonance frequency of 600 MHz essentially as described [22]. For each sample two spectra were acquired focusing on the aliphatic and aromatic regions. For the aliphatic spectra, a matrix of 1024 × 1500 (f2 × f1) complex data points was acquired and zero-filled to 4096 complex data points in f1. The spectral widths were 6000 Hz and 5100 Hz in the 1H- and 13C-dimensions, respectively. The narrow spectral width in the 13C-dimension leads back-folding of part of the signals to the empty regions of the spectrum. For the aromatic region, a matrix of 1024 × 800 complex data points was acquired and zero-filled to 2048 complex data points in f1. The spectral widths for the aromatic spectra were 6000 Hz and 2815 Hz in the 1H- and 13C-dimensions, respectively. All spectra were weighted with a cosine function in both dimensions prior to the Fourier transformation. The spectra were processed using the standard Varian spectrometer software VNMR (version 6.1, C).
Metabolic Flux Ratio (METAFoR) analysis
The software FCAL (R.W. Glaser; FCAL 2.3.1) [25] was used for the integration of 13C-scalar fine structures of proteinogenic amino acid carbon signals in the 13C-HSQC NMR spectra and the calculation of relative abundances of intact carbon fragments originating from a single source molecule of glucose. The nomenclature used here for the intact carbon fragments, fragmentomers, has been described previously [22]. Briefly, f(1) represents the fraction of molecules in which the observed carbon atom and the neighboring carbons originate from different source molecules of glucose, f(2) the fraction of molecules in which the observed carbon atom and one of the two neighboring atoms originate from the same source molecule of glucose, and f(3) the fraction of molecules in which the observed carbon atom and both neighboring carbons originate from the same source molecule of glucose. If the observed carbon exhibits significantly different 13C-13C scalar coupling constants with the neighboring carbons, f(2) and f(2*) can be distinguished. The fraction of molecules with a conserved bond between the observed carbon atom and the neighboring carbon with the smaller coupling is represented by f(2). f(2*) then denotes the fraction of molecules where the bond is conserved between the observed carbon and the neighboring carbon with the larger coupling. If the observed carbon is located at the end of a carbon chain, f(1) and f(2) fragmentomers can be observed indicating the conservation of the two terminal carbon fragment of the molecule.
The degree of13C-labeling of the biomass amino acids was determined from the 13C-scalar fine structures of Leu-Cβ and Val-Cγ2. The biomass was assumed to be fully produced from the fractionally labelled carbon source in the glucose experiments and in sorbitol experiments without sophorose induction because the dry weight of the inoculum was negligible. For the sorbitol experiments with sophorose induction the fraction of labeled biomass was estimated from the dry weight measurement (data not shown). The model of the central carbon metabolism network used in the METAFoR analysis was the one previously developed for eukaryotic metabolism of the yeast S. cerevisiae [26] (Figure 6). Fragmentomer information obtained from proteinogenic amino acids can be traced back to the metabolic intermediates in central carbon metabolism through the amino acid synthesis pathways to assess ratios of intracellular fluxes which merge at a metabolic network junction [26]. The biosynthetic pathways of amino acids in T. reesei were reconstructed with carbon path analysis method ReTrace [21] described in the next section.

Metabolic network model. Eukaryotic central carbon metabolism network model [26].
Carbon path analysis
The reconstruction of amino acid biosynthetic pathways from their precursors in T. reesei was performed with ReTrace. ReTrace is a recent computational pathway analysis method [21], which can be queried to discover branching metabolic pathways in a universal metabolic database. Specifically, ReTrace aims to find pathways which transfer as many atoms from source to target metabolites as possible.
The reaction database used in ReTrace analysis was KEGG LIGAND, downloaded in March 2009 [40]. Reaction database contained 7827 reactions and 15400 compounds. Atom map**s, that describe how atoms are transferred in a reaction from substrate to product metabolites, were defined for 33795 substrate-product pairs in the RPAIR database, which is a subdatabase of KEGG. All reactions were considered bidirectional. To compute reaction scores, a database consisting of 101136 sequences annotated with an EC number in UniProt version 9.3 [41] was queried with the 9129 protein sequences in T. reesei genome [Localization of amino acid biosynthetic enzymes in T. reesei TargetP, a machine learning method based on neural networks, which predicts both chloroplast and mitochondrial targeting peptides and secretory signal peptides, was utilized to predict the probable subcellular localization of some amino acid biosynthetic enzymes in T. reesei [33, 34]. The prediction performance of non-plant mitochondrial targeting peptides with TargetP has been measured to be 80 - 90% sensitivity and 70% specificity [33]. TargetP reported, for each analyzed peptide sequence, the probability that the peptide contained some signal peptide (SP), a mitochondrial targeting peptide (mTP) or cytosolic targeting peptide (cTP) presequence. In addition, a numerical reliability class (RC) between 1 and 5 was reported. The reliability class was derived from the difference of highest and second-highest probabilities assigned to the prediction classes SP, mTP, cTP or "other". The class "other" indicates the probability that no subcellular location sorting signal was found. If the difference was greater than 0.8, RC equals 1; if the difference was below 0.2, RC equals 5.