
Abstract
Background
Expressed sequence tags (ESTs) have been used for rapid gene discovery in a variety of organisms and provide a valuable resource for whole genome annotation. Although the genome of one marsupial, the opossum Monodelphis domestica, has now been sequenced, no EST datasets have been reported from any marsupial species. In this study we describe an EST dataset from the bandicoot, Isoodon macrourus, providing information on the transcriptional profile of the bandicoot thymus and the opportunity for a genome wide comparison between the bandicoot and opossum, two distantly related marsupial species.
Results
A set of 1319 ESTs was generated from sequencing randomly chosen clones from a bandicoot thymus cDNA library. The nucleic acid and deduced amino acid sequences were compared with sequences both in GenBank and the recently completed whole genome sequence of M. domestica. This study provides information on the transcriptional profile of the bandicoot thymus with the identification of genes involved in a broad range of activities including protein metabolism (24%), transcription and/or nucleic acid metabolism (10%), metabolism/energy pathways (9%), immunity (5%), signal transduction (5%), cell growth and maintenance (3%), transport (3%), cell cycle (0.7%) and apoptosis (0.5%) and a proportion of genes whose function is unknown (5.8%). Thirty four percent of the bandicoot ESTs found no match with annotated sequences in any of the public databases. Clustering and assembly of the 1319 bandicoot ESTs resulted in a set of 949 unique sequences of which 375 were unannotated ESTs. Of these, seventy one unannotated ESTs aligned to non-coding regions in the opossum, human, or both genomes, and were identified as strong non-coding RNA candidates. Eighty-four percent of the 949 assembled ESTs aligned with the M. domestica genome sequence indicating a high level of conservation between these two distantly related marsupials.
Conclusion
This study is among the first reported marsupial EST datasets with a significant inter-species genome comparison between marsupials, providing a valuable resource for transcriptional analyses in marsupials and for future annotation of marsupial whole genome sequences.
Similar content being viewed by others

Background
Marsupials and eutherian mammals are each other's closest relatives, having diverged from a common ancestor around 172 million years ago (MYA) [1–3]. Marsupials are most clearly distinguished from eutherians by their mode of reproduction, giving birth to young at a relatively immature stage of development and placing greater emphasis on lactation than on intrauterine development during pregnancy [4]. Much of the development that occurs in utero in eutherians is postnatal in marsupials, including the development of the immune system [5–9]. However, young marsupials are most vulnerable to dying not at birth, but at the time of emergence from the nest or pouch, suggesting that maternal antibodies and anti-microbials provide protection during the lactation period [4].
Although many marsupial genes have been identified using homology based approaches such as PCR and DNA-DNA hybridization, the low conservation of some genes makes them difficult to amplify and clone based solely on sequence similarity with homologs from other species [10–13]. Expressed sequence tags (ESTs) are single-pass, partial sequences of cDNA clones and have been used extensively for gene discovery and genome map** in humans and other organisms [20]. All marsupials develop a simple yolk sac placenta during pregnancy. In addition to the yolk sac placenta, bandicoots are unique among marsupials in that they are the only group that develops a highly invasive chorio-allantoic placenta, similar to that of eutherian mammals, during the final 2 days of their 12.5 day gestation period. The post-natal growth of bandicoots is the most rapid of any marsupial, suggesting that the chorio-allantoic placenta is likely a more efficient organ of exchange than the yolk sac placenta [21]. Another unique characteristic of bandicoots is the process of chromosome inactivation. In most somatic tissues during development the paternally derived X chromosome in females and the Y chromosome in males is eliminated [22].
In an effort to identify immune related genes in the bandicoot, we carried out an analysis of ESTs using a thymus cDNA library from the northern brown bandicoot, I. macrourus. Due to its role in central tolerance, the thymus expresses a large percentage of the proteome and therefore allows for the identification of a broad range of genes including those involved in the immune response [23]. This study represents one of the first analyses of ESTs reported from any marsupial. In addition to providing important information on the gene profile of the bandicoot thymus, comparison of the bandicoot ESTs with the opossum genome is among the largest genome wide comparison between two marsupials reported to date. This information provides valuable resources for future studies of gene expression in bandicoots and other marsupials and will assist in the annotation of marsupial whole genome sequences.
Results and discussion
Overview of expressed sequence tags
The 5' ends of 2000 randomly selected cDNA clones from the bandicoot thymus cDNA library were sequenced, yielding a total of 1319 ESTs. The average insert size of the bandicoot cDNA clones was 1.4 Kb and the average length of useable sequence obtained was 412 bp. By comparison with sequences in the public databases, 798 (60%) of the bandicoot ESTs had homology to known mammalian genes and were placed into several categories according to their putative biological functions (Figure 1). These included tissue specific transcripts encoding proteins involved in immune function and those that are ubiquitously expressed and involved in broad functions such as development and cell maintenance. A further 76 sequences (5.8%) matched previously identified coding regions of unknown function and 445 sequences (34%) resulted in no match with annotated sequences in any of the public databases (Figure 1).

The distribution of 1319 expressed sequence tags (ESTs) from a bandicoot thymus cDNA library.
ESTs matching genes with known function
Figure 1 shows the distribution of transcripts identified in the bandicoot thymus. Not surprisingly, the most highly expressed transcripts were housekee** genes necessary for cell maintenance within all tissues, including the thymus. As in many other EST studies, transcripts involved in protein metabolism were the most abundant (24%), followed by those involved in transcription and/or nucleic acid metabolism (10%) and metabolism/energy pathways (9%) (Figure 1). While many transcripts were identified only once (310 out of 798), 120 were detected between 2–22 times. A total of 430 different genes were identified among the 1319 clones analyzed, to our knowledge making it the largest current set of annotated genes for a Peramelid marsupial. Ribosomal proteins (assorted 40S and 60S) were the most redundant sequences identified, making up 221 ESTs (see Additional file 1). This is similar to the expression of ribosomal proteins in other EST libraries from the human thymus and other tissues [24]. Other highly expressed housekee** genes encoded proteins necessary for intracellular protein trafficking, cell division and substance transport including cytochrome c oxidase (23 ESTs), histones (21 ESTs), NADH dehydrogenase (18 ESTs) and ubiquitin (15 ESTs) (see Additional file 1).
Since the function of the thymus is the maturation and selection of T cells, we were interested in the immune related ESTs among the bandicoot dataset. The thymus is important in the development of T cell tolerance to self major histocompatibility complex (MHC), including class Ib molecules with tissue specific distribution patterns such as those involved in fetal-maternal tolerance. Although the cDNA library used in the present study was from a juvenile male bandicoot, molecules involved in allorecognition are generally expressed in thymuses from both males and females, likely reflecting their role in immune regulation in addition to the fetal-maternal interface. The thymus therefore provides an overview of immune genes relevant to both general immunity and to mechanisms of immune tolerance.
Seventy of the 1319 ESTs showed homology to genes associated with immune function and/or T cell development (see Additional File 1). The expression profile of immune related genes in the bandicoot thymus is consistent with current knowledge of thymic T cell maturation in other mammals. T and B cell receptors and co-receptors, antigen processing genes, cytokine receptors and genes encoding molecules involved in innate immune function were among the immune related molecules identified among the bandicoot thymus EST dataset (Table 1).
The identification of immunoglobulin and CD79a transcripts indicates that B cells are present in the bandicoot thymus. This is consistent with previous reports indicating that B cells are present in the thymus of eutherians and marsupials. CD79a-positive cells have been observed in the thymuses of adult and develo** marsupials [8, 25, 26]. In addition, the thymuses of neonatal and adult mice and adult humans contain a small population of B lymphocytes [27–29]. Nango et al. (1991) [29] have suggested that thymic B cells in humans may play a role in negative selection of T cells. It is likely their role is similar in marsupials.
Functional studies suggest that marsupials display some interesting differences in their immune response compared to eutherian mammals. For instance, marsupials are capable of rejecting allogenic skin grafts in a manner similar to that of eutherians, however, the average time for complete rejection appears to be significantly longer than the time observed for other mammalian species. In addition, weak or non-existent mixed lymphocyte responses and a poor capacity for immunoglobulin class switching have been reported for several marsupial species. Some marsupials also show increased susceptibility to pathogens such as Mycobacterium tuberculosis and Trypansoma cruzi (reviewed in [30]). Further information on the molecules involved in the marsupial immune response will assist in determining the mechanisms responsible for these differences.
A variety of immune related molecules not previously cloned from any marsupial were identified among the bandicoot ESTs. Amongst these were ESTs with homology to antigen processing genes including proteasome subunits (PSMB8, PSMB9, PSMB10 and PA28α) and the MHC associated class II invariant chain. Of the four cytokine receptor genes identified in the bandicoot thymus (TNFR, LTBR, IL-18R and IFNGR), only IFNGR has been previously identified in the whole genome sequence of M. domestica and none of these receptors have been cloned previously from a marsupial [31]. Four genes involved in the innate immune system were among the bandicoot thymus ESTs that have not been previously reported from a marsupial: complement component 3 (C3), proteoglycan core protein, glioma pathogenesis related protein 1 (GliPR1) and a homolog of a cathelicidin antimicrobial peptide (Table 1). C3 is the central component of the complement cascade important in innate immunity and inflammation. Homologs of complement components including C3 have been identified in a range of vertebrates and invertebrates providing evidence for an ancient origin of this system [32]. Proteoglycan core protein and GliPR1 are novel innate immune molecules not previously described in any marsupial. Proteoglycan core protein is the primary proteoglycan of cytotoxic granules and is involved in granule mediated apoptosis [33]. GliPR1 belongs to a group of plant pathogenesis-related proteins and is believed to play a role in the innate immune response of vertebrates [34]. The bandicoot cathelicidin is the first identified in any marsupial to date. Cathelicidins have been identified in a range of mammals and more recently in hagfish and rainbow trout [35, 36]. Potential antimicrobial compounds have been identified in the pouches of koalas but none were homologous to known cathelicidins [37]. Such anti-microbial compounds likely provide an innate layer of protection for the vulnerable pouch-young prior to the development of their adaptive immune system. The identification of a variety of immune related molecules in the present study provides evidence that the complexity of the marsupial immune system is comparable to that of eutherian mammals despite the differences in immune responses previously reported.
Comparison of bandicoot ESTs with the whole genome sequence of the opossum
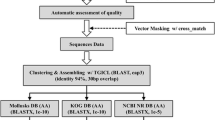
The availability of a whole genome sequence from M. domestica allows for a significant comparison between two marsupial genomes. To better characterize the homology between the bandicoot EST sequences and the opossum genome, the 1319 bandicoot ESTs were clustered and assembled using EGassembler [38], resulting in 144 contigs and 805 singletons. Assembled ESTs were used for the cross species comparison to reduce the influence of high copy numbers of abundantly expressed transcripts in the raw EST dataset. Thirty EST sequences were discarded for being too short after masking and trimming. Contigs ranged in size from 254 to 1189 bp, with a median size of 546 bp. The largest cluster contained 11 transcripts and corresponds to 60S ribosomal protein L26.
Of the 949 assembled bandicoot ESTs, 797 (84%) found a match with sequence from the opossum genome (Table 2). By comparison, Rink et al. (2006) [39] reported that 1422 of 2035 pig ESTs (70%) found a match in the human genome. The divergence between artiodactyls and humans is estimated to have occurred around 80–100 MYA [40], similar to bandicoots and opossums which last shared a common ancestor approximately 75 MYA [40–42]. Given the time period separating bandicoots and opossums, the large proportion of bandicoot sequences that aligned with the opossum genome sequence is remarkable. The smaller dataset of bandicoot ESTs may have caused a bias towards the identification of highly expressed, highly conserved housekee** genes, therefore resulting in a greater number of matches with sequence in the opossum genome sequence.
There were 152 assembled ESTs that did not align with the whole genome sequence of the opossum at either the nucleotide or amino acid level (Table 2). Not surprisingly, the majority of these ESTs (134) also had no hit with annotated sequences from the public databases and are included below in the analysis of non-coding RNAs (ncRNAs). Of the remaining 18, 15 matched proteins of known function and three matched sequences of unknown function in other species. It is possible that some of these genes are absent from the current opossum genome assembly due to unsequenced gaps. Alternatively, some genes may have diverged rapidly or been lost in the opossum lineage.
Non-coding RNAs
Analyses of EST datasets from other species have reported a proportion of sequences that represent ncRNAs [43]. ncRNAs make up a significant proportion of the mammalian transcriptome and play a variety of roles, including transcriptional regulation, chromosome replication, RNA processing and modification, mRNA stability and translation, and protein degradation and translocation [44–46]. ncRNAs range from the 22 nucleotide family of microRNAs (miRNAs) to the 100–200 nucleotide small RNAs (sRNAs) commonly found as translational regulators in bacterial cells to >10000 nucleotides for RNAs involved in gene silencing in higher eukaryotes [46].
After assembly, the 445 ESTs that did not match an annotated sequence in the public databases yielded 375 unique sequences (consisting of 144 contigs and 231 singletons). These ranged from 106 to 1019 bp in length. Some of the shorter clones in this category may contain insufficient sequence to identify coding regions and many of these sequences may represent the 3' or 5' untranslated region (UTR) of genes. To determine whether open reading frames (ORFs) were present in any of the unmatched ESTs, the web-based program, ESTScan was used to scan these ESTs for potential ORFs [47]. This resulted in the identification of putative ORFs in only 14 of the 375 unmatched ESTs. These 14 ORFs ranged in length from 84–383 bp. It is possible that these sequences encode proteins that have not been identified in other species or they may be composed of sequences too divergent to be recognized by sequence comparison programs. To identify divergent gene homologs among the 375 unmatched ESTs, the six-frame translation was also searched using all profiles in the Pfam database using a hidden Markov model search program (HMMER), however no significant matches were identified.
We investigated the possibility that some of the 375 unmatched bandicoot ESTs are conserved ncRNAs by comparing them to the opossum and human genome sequences (Figure 2; see Additional file 2) and their ENSEMBL or UCSC annotations. ESTs that aligned more than 5 kb from an annotated gene were labeled High Quality Non-Coding (HQNC) sequences. Of these 375 unmatched ESTs, 158 were considered unlikely ncRNA candidates because they either aligned to the opossum genome alone (138 clones) or the human genome alone (1 clone) or both (19 clones) and were within 5 kb of an annotated gene. These may be UTRs from coding mRNAs. Pre-miRNA and small nucleolar RNAs (snoRNAs) may also be represented in this group, as many miRNAs and snoRNAs are encoded in introns [48]. However, searching this set with the sequences of known pre-miRNAs and snoRNAs did not result in any significant matches (e-value ≤ 10-4). A further 13 ESTs aligned to both the human and opossum genomes, but were within 5 kb of an annotated gene in only one of the species. Thus, these ESTs obtained inconsistent HQNC status and are unlikely to be ncRNAs. Of the remaining unmatched ESTs, 133 aligned to neither the opossum nor human genomes, and are therefore not conserved. These may still be functional ncRNAs, since larger non-sno, non-micro ncRNAs (for example XIST and AIR) are poorly conserved and are thought to be under the influence of different evolutionary constraints [49]. The remaining 71 unmatched ESTs represent an integrated set of High Quality Non-Coding (iHQNC) sequences from comparison with two genomes and are therefore strong candidates for ncRNA. Of these 71 iHQNC sequences, 67 ESTs aligned to the opossum genome sequence only and may be marsupial specific ncRNAs (Fig. 2; see Additional file 2). Four ESTs aligned to both the human and opossum genomes and may represent ncRNAs conserved across all therian mammals. The function of both the marsupial specific and conserved ncRNAs identified in this study remains to be determined. However, since ncRNAs co-evolve with the genes they regulate it is not surprising that some are marsupial specific.

The distribution of results from aligning 375 unannotated assembled ESTs with the opossum and human genomes. An Integrated High Quality Non Coding (iHQNC) set of ESTs that aligned more than 5 kb from an annotated feature were identified. ESTs that aligned to HQNC regions in one genome but were within 5 kb of an annotated gene in the other genome are labeled "inconsistent" HQNC.
The 71 bandicoot iHQNC sequences represent 7.5% of the clustered and assembled EST dataset. By comparison, 7% of 60770 full length mouse cDNAs and 1.9% of a set of 19626 chicken EST contigs were identified as ncRNAs [43, 45]. Thus, the results of the bandicoot EST analysis is comparable to that of the mouse and may be representative of the expression of ncRNAs in mammalian tissues.
Conclusion
In summary, this study provides an overview of transcripts expressed in the bandicoot thymus, providing the opportunity for a genome wide comparison with the M. domestica whole genome sequence. Comparison of the bandicoot EST dataset with the opossum whole genome sequence revealed a high level of homology between the genomes of these two distantly related marsupials. This study has also expanded the number of immunity related genes known for marsupials, providing the first step in develo** the tools necessary for further studies of immune function in marsupials. Overall, these results demonstrate the utility of an EST approach for gene discovery in marsupials and provide a useful resource of immune and non-immune related genes for transcriptional analysis in marsupials. Furthermore, the identification of genes specifically in the bandicoot that are likely to play a role in maternal-fetal interactions, will greatly enhance this species as a model organism. Such genes include those involved in allorecognition, such as genes of the MHC and TCR, as well as genes with a role in innate immunity, like the cathelicidin antimicrobial peptide.
Methods
Isolation and analysis of Expressed Sequence Tag (EST) clones
The thymus cDNA library and the isolation of ESTs have been described previously [50]. Briefly, two thousand randomly selected colonies were sequenced from a mass excision of the unamplified bandicoot thymus cDNA library using the T3 primer. Sequences were compared to sequences in the protein and nucleotide databases (Swissprot, nt and nr) in GenBank and the M. domestica whole genome sequence using the BLAST algorithms [51]. Only matches with e-values of ≤10-5 were considered significant. Those sequences that consisted of vector sequence only, bad sequence reads or that were less than 100 nucleotides in length were excluded from the analysis. The web-based, ESTScan was used to scan sequences that showed no match with any of the public databases to identify whether they contained potential ORFs [47]. Sequences were categorized into groups according to their predicted biological function, following the classifications of The Gene Ontogeny Consortium (GO) [52] and human protein reference database [53]. The ESTs generated in this study have been submitted to GenBank (accession nos. EE743888–EE745206).
Comparison with M. domestica genome sequence
To better characterize the homology between the bandicoot and the opossum, the ESTs were masked for mammalian repeats using RepeatMasker and then clustered and assembled using EGassembler [38]. Masking of repeats and organelle sequence was disabled in EGassembler.
The whole genome of the South American opossum, M. domestica, has been sequenced by the Broad Institute (MIT). All analyses were performed using MonDom4 (GenBank accession number AAFR03000000). The clustered and assembled ESTs were compared with the whole genome sequence of the opossum using BLASTN and TBLASTX [51]. Only matches with e-values ≤10-4 were considered significant.
To identify diverged homologs in the unannotated ESTs, the 6 frame translation was searched with all Pfam profile hidden Markov models [54, 55] using HMMer [56]. The unannotated ESTs were then aligned with predicted peptide sequences from the ENSEMBL M. domestica genebuild (Build 39) using BLASTX.
Detection of conserved non-coding RNAs
To create a set of High Quality Non-Coding (HQNC) ESTs [43] we aligned the 375 ESTs that showed no match to annotated genes in the public database to the M. domestica genome (MonDom3, released January 2006) using BLASTN and TBLASTX. The best matches with e-values ≤ 10-4 were compared with the Ensembl annotation (Build 39). Since UTRs are essentially unrepresented in this annotation, ESTs less than 5 kb from an ENSEMBL feature were considered to be possible UTRs. The remaining ESTs formed a HQNC set.
To make use of the higher quality annotation of the human genome, we also aligned the 375 unmatched ESTs to the human genome (NCBI Build 36.1, release March 2006). The best matches (e-value ≤ 10-4) were compared with the UCSC genome browser 'known gene' annotation (hg18), which is based on protein data from UniProt and mRNA data from the NCBI RefSeq collection and GenBank. Once again ESTs that were within 5 kb of an annotated gene were considered to be possible UTRs. This provided a second HQNC set based on a higher quality annotation, but using a more distantly related species.
The bandicoot HQNC ESTs identified from alignment to the opossum or human genomes were then combined into an integrated High Quality Non-coding (iHQNC) EST set. ESTs that aligned to both the opossum and human genomes and were members of the HQNC sets in both species were regarded as conserved HQNC and included in the iHQNC set. ESTs that aligned to only one genome and were present in the HQNC set for that species were also included as iHQNCs. ESTs that aligned to both genomes and were within 5 kb of an annotated gene in one of the species but not the other were regarded as having inconsistent HQNC status. The resulting iHQNC set thus contains conserved HQNC sequences and HQNC sequences that are highly diverged or putatively lost from one species or the other.
Finally, we compared the unmatched ESTs with known human snoRNAs from the snoRNA-LBME-db [57] and with predicted stem-loop sequences (which include the pre-miRNA) from miRBase [58, 59] using BLASTN (e-value ≤ 10-4).
Abbreviations
- EST:
-
Expressed sequence tag
- HQNC:
-
High quality non coding
- iHQNC:
-
Integrated high quality non coding
- ORF:
-
Open reading frame
- MHC:
-
Major Histocompatibility Complex
- MYA:
-
million years ago
- miRNA:
-
microRNA
- ncRNA:
-
non coding RNA
- PSMB:
-
Proteasome subunit beta
- sRNA:
-
small RNA
- snoRNA:
-
small nucleolar RNA
- TCR:
-
T cell receptor
- UTR:
-
Untranslated region
References
Baker ML, Harrison GA, Wares JP, Miller RD: The relationship of the marsupial families and the mammalian subclasses based on recombination activating gene-1. J Mammal Evol. 2004, 11: 1-16. 10.1023/B:JOMM.0000029143.39776.ec.
Belov K, Harrison GA, Miller RD, Cooper DW: Molecular cloning of four lambda light chain cDNAs from the Australian brushtail possum (Trichosurus vulpecula). Eur J Immunogenet. 2002, 29: 95-99. 10.1046/j.1365-2370.2002.00286.x.
Janke A, Xu X, Arnason U: The complete mitochondrial genome of the wallaroo (Macropus robustus) and the phylogenetic relationship among Monotremata, Marsupialia, and Eutheria. Proc Natl Acad Sci USA. 1997, 94: 1276-1281. 10.1073/pnas.94.4.1276.
Tyndale-Biscoe CH, Renfree M: Reproductive physiology of marsupials. 1987, London: Cambridge University Press
Ashman RB, Papadimitriou JM: Development of lymphoid tissue in a marsupial, Setonix brachyurus (quokka). Acta Anat. 1975, 91: 594-611.
Hubbard GB, Saphire DG, Hackleman SM, Silva MV, Vandeberg JL, Stone WH: Ontogeny of the thymus gland of a marsupial (Monodelphis domestica). Lab Anim Sci. 1991, 41: 227-232.
Basden K, Cooper DW, Deane EM: Development of the lymphoid tissues of the tammar wallaby Macropus eugenii. Reprod Fertil Dev. 1997, 9: 243-254. 10.1071/R96032.
Baker ML, Gemmell E, Gemmell RT: Ontogeny of the immune system of the brushtail possum, Trichosurus vulpecula . Anat Rec. 1999, 256: 354-365. 10.1002/(SICI)1097-0185(19991201)256:4<354::AID-AR3>3.0.CO;2-7.
Cisternas PA, Armati PJ: Development of the thymus, spleen, lymph nodes and liver in the marsupial, Isoodon macrourus (Northern brown bandicoot, Peramelidae). Anat Embryol. 1999, 200: 433-443. 10.1007/s004290050293.
Miska KB, Miller RD: Marsupial Mhc class I: classical sequences from the opossum, Monodelphis domestica. Immunogenetics. 1999, 50: 89-93. 10.1007/s002510050692.
Baker ML, Rosenberg GH, Zuccolotto P, Harrison GA, Deane EM, Miller RD: Further characterization of T cell receptor chains of marsupials. Dev Comp Immunol. 2001, 25: 495-507. 10.1016/S0145-305X(01)00016-7.
Harrison GA, Young LJ, Watson CM, Miska KB, Miller RD, Deane EM: A survey of type I interferons from a marsupial and monotreme: implications for the evolution of the type I interferon gene family in mammals. Cytokine. 2003, 21: 105-119. 10.1016/S1043-4666(03)00029-2.
Baker ML, Belov K, Miller RD: Unusually similar patterns of antibody V segment diversity in distantly related marsupials. J Immunol. 2005, 174: 5665-5671.
Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, **ao H, Merril CR, Wu A, Olde B, Moreno RF, Kerlavage AR, McCombie WR, Venter JC: Complementary DNA Sequencing: Expressed Sequence Tags and Human Genome Project. Science. 1991, 252: 1651-1656. 10.1126/science.2047873.
Smith LC, Chang L, Britten RJ, Davidson EH: Sea urchin genes expressed in activated coelomocytes are identified by expressed sequence tags: Complement homologues and other putative immune response genes suggest immune system homology within the deuterostomes. J Immunol. 1996, 156: 593-602.
Suzuki T, Shin IT, Kohara Y, Kasahara M: Transcriptome analysis of hagfish leukocytes: a framework for understanding the immune system of jawless fishes. Dev Comp Immunol. 2004, 28: 993-1003. 10.1016/j.dci.2004.04.005.
Nair SV, Del Valle H, Gross PS, Terwilliger DP, Smith LC: Macroarray analysis of coelomocyte gene expression in response to LPS in the sea urchin. Identification of unexpected immune diversity in an invertebrate. Physiol Genomics. 2005, 22: 33-47. 10.1152/physiolgenomics.00052.2005.
Yu J, Farjo R, MacNee SP, Baehr W, Stambolian DE, Swaroop A: Annotation and analysis of 10,000 expressed sequence tags from develo** mouse eye and adult retina. Genome Biol. 2003, 4: R65-10.1186/gb-2003-4-10-r65.
Zhang B, ** W, Zeng Y, Su Z, Hu S, Yu J: EST-based analysis of gene expression in the porcine brain. Genomics Proteomics Bioinformatics. 2004, 2 (4): 237-244.
Tyndale-Biscoe CH: Life of Marsupials. 2005, Collingwood: CSIRO Publishing
Padykula HA, Taylor JM: Marsupial placentation and its evolutionary significance. J Reprod Fertil Suppl. 1982, 31: 95-104.
Johnston PG, Watson CM, Adams M, Paull DJ: Sex chromosome elimination, X chromosome inactivation and reactivation in the southern brown bandicoot Isoodon obesulus (Marsupialia: Peramelidae). Cytogenet Genome Res. 2002, 99: 119-124. 10.1159/000071583.
Kyewski B, Klein L: A central role for central tolerance. Annu Rev Immunol. 2006, 24: 571-606. 10.1146/annurev.immunol.23.021704.115601.
Hwang M-Y, Kang Y-J, Kim Y-H, Scherer SW, Tsui L-C, Sohn U: Generation and chromosome map** of expressed sequence tags (ESTs) from a human infant thymus. Genome. 1999, 42: 457-464. 10.1139/gen-42-3-457.
Hemsley SW, Canfield PJ, Husband AJ: Immunohistological staining of lymphoid tissue in four Australian marsupial species using species cross-reactive antibodies. Immunol Cell Biol. 1995, 73: 321-325.
Canfield P, Hemsley S, Connolly J: Histological and immunological study of the develo** and involuting superficial cervical thymus in the koala (Phascolarctos cinereus). J Anat. 1996, 189: 159-169.
Isaacson PG, Norton AJ, Addis BJ: The human thymus contains a novel population of B lymphocytes. Lancet. 1987, 2: 1488-1490. 10.1016/S0140-6736(87)92622-5.
Miyama-Inaba M, Kuma S, Inaba K, Ogata H, Iwai H, Yasumizu R, Muramatsu S, Steinman RM, Ikehara S: Unusual phenotype of B cells in the thymus of normal mice. J Exp Med. 1988, 168: 811-816. 10.1084/jem.168.2.811.
Nango K, Inaba M, Inaba K, Adachi Y, Than S, Ishida T, Kumamoto T, Uyama M, Ikehara S: Ontogeny of thymic B cells in normal mice. Cell Immunol. 1991, 133: 109-115. 10.1016/0008-8749(91)90183-C.
Old JM, Deane EM: Development of the immune system and immunological protection in marsupial pouch young. Dev Comp Immunol. 2000, 24: 445-454. 10.1016/S0145-305X(00)00008-2.
Wong ESW, Young LJ, Papenfuss AT, Belov K: In silico identification of opossum cytokine genes suggests the complexity of the marsupial immune system rivals that of eutherian mammals. Immunome Res. 2006, 2: 4-10.1186/1745-7580-2-4.
Nonaka M, Yoshizaki F: Evolution of the complement system. Mol Immunol. 2004, 40: 897-902. 10.1016/j.molimm.2003.10.009.
Murphy EV, Zhang Y, Zhu W, Biggs J: The human glioma pathogenesis-related protein is structurally related to plant pathogenesis-related proteins and its gene is expressed specifically in brain tumors. Gene. 1995, 159: 131-135. 10.1016/0378-1119(95)00061-A.
Metkar SS, Wang B, Aguilar-Santelises M, Raja SM, Uhlin-Hansen L, Podack E, Trapani JA, Froelich CJ: Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-Serglycin complexes into target cells without plasma membrane pore formation. Immunity. 2002, 16: 417-428. 10.1016/S1074-7613(02)00286-8.
Uzzell T, Stolzenberg ED, Shinnar AE, Zasloff M: Hagfish intestinal antimicrobial peptides are ancient cathelicidins. Peptides. 2003, 24: 1655-1667. 10.1016/j.peptides.2003.08.024.
Chang C-I, Pleguezuelos O, Zhang Y-A, Zou J, Secombes CJ: Identification of a Novel Cathelicidin Gene in the Rainbow Trout, Oncorhynchus mykiss . Infect Immun. 2005, 73: 5053-5064. 10.1128/IAI.73.8.5053-5064.2005.
Bobek G, Deane EM: Possible antimicrobial compounds fromthe pouch of the koala, Phascolarctos cinereus . Lett Peptide Sci. 2002, 8: 133-137.
Masoudi-Nejad A, Tonomura K, Kawashima S, Moriya Y, Suzuki M, Itoh M, Kanehisa M, Endo T, Goto S: EGassembler: Online bioinformatics service for large-scale processing, clustering and assembling ESTs and genomic DNA fragments. Nuc Acid Res. 2006, 34 (Web Server): w459-62. 10.1093/nar/gkl066.
Rink A, Eyer K, Roelofs B, Priest KJ, Sharkey-Brockmeier KJ, Leknong S, Karajusuf EK, Bang J, Yerle M, Milan D, Liu WS, Beattie CW: Radiation hybrid map of the porcine genome comprising 2035 EST loci. Mamm Genome. 2006, 17: 878-885. 10.1007/s00335-005-0121-0.
Springer MS, Murphy WJ, Eizirik E, O'Brien SJF: Placental mammal diversification and the Cretaceous-Tertiary boundary. Proc Natl Acad Sci USA. 2003, 100: 1056-1061. 10.1073/pnas.0334222100.
Kirsch JA, Lapointe FJ, Springer MS: DNA-hybridization studies of marsupials and their implications for Metatherian classification. Aust J Zool. 1997, 45: 211-280. 10.1071/ZO96030.
Nilsson MA, Arnason U, Spencer PBS, Janke A: Marsupial relationships and a timeline for marsupial radiation in South Gondwana. Gene. 2004, 340: 189-196. 10.1016/j.gene.2004.07.040.
Hubbard SJ, Grafham DV, Beattie KJ, Overton IM, McLaren SR, Croning MDR, Boardman PE, Bonfield JK, Burnside J, Davies RM, Rarrell ER, Francis MD, Griffiths-Jones S, Humphray SJ, Hyland C, Scott CE, Tang H, Taylor RG, Tickle C, Brown WRA, Birney E, Rogers J, Wilson SA: Transcriptome analysis for the chicken based on 19,626 finished cDNA sequences and 485,337 expressed sequence tags. Genome Res. 2004, 15: 174-183. 10.1101/gr.3011405.
Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, Kimura K, Makita H, Sekine M, Obayashi M, Nishi T, Shibahara T, Tanaka T, Ishii S, Yamamoto J, Saito K, Kawai Y, Isono Y, Nakamura Y, Nagahari K, Murakami K, Yasuda T, Iwayanagi T, Wagatsuma M, Shiratori A, Sudo H, et al.: Complete sequencing and characterization of 21,243 full-length human cDNAs. Nature Genet. 2004, 36: 40-45. 10.1038/ng1285.
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, et al.: Analysis of the mouse transcriptome based on functional annotation of 60,770 full length cDNAs. Nature. 2002, 420: 563-573. 10.1038/nature01266.
Storz G: An expanding universe of noncoding RNAs. Science. 2002, 296: 1260-1263. 10.1126/science.1072249.
Iseli C, Jongeneel CV, Bucher P: ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999, 138-148.
Mattick JS, Makunin IV: Non-coding RNA. Hum Mol Genet. 2006, 15: R17-29. 10.1093/hmg/ddl046.
Pang KC, Frith MC, Mattick JS: Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 22: 1-5. 10.1016/j.tig.2005.10.003.
Baker ML, Osterman AK, Brumburgh S: Divergent T cell receptor delta chains from marsupials. Immunogenetics. 2005, 57: 665-673. 10.1007/s00251-005-0030-0.
Altschul SF, Gish W, Miller W, Myers EM, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215 (3): 403-410.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet. 2000, 25: 25-29. 10.1038/75556.
Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, Chandrika KN, Deshpande N, Suresh S, Rashmi BP, Shanker K, Padma N, Niranjan V, Harsha HC, Talreja N, Vrushabendra BM, Ramya MA, Yatish AJ, Joy M, Shivashankar HN, Kavitha MP, Menezes M, Choudhury DR, Ghosh N, Saravana R, Chandran S, Mohan S, Jonnalagadda CK, Prasad CK, et al.: Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13: 2363-2371. 10.1101/gr.1680803.
Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer ELL, Bateman A: Pfam: clans, web tools and services. Nucleic Acids Res. 2006, 34: D247-D251. 10.1093/nar/gkj149.
Pfam (Wellcome Trust Sanger Institute). [http://www.sanger.ac.uk/Software/Pfam/]
Hmmer. [http://hmmer.janelia.org/]
Lestrade L, Weber MJ: snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006, 34: D158-162. 10.1093/nar/gkj002.
Griffiths-Jones S: The microRNA Registry. NAR, Database Issue. 2004, 32: D109-D111.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ: miRBase: microRNA sequences, targets and gene nomenclature. NAR, Database Issue. 2006, 34: D140-44.
Acknowledgements
This publication was made possible by National Institutes of Health grant Number IP20RR18754 from the Institutional Development Award (IDeA) program of the National Center for Research Resources (MLB and RDM) and a National Science Foundation award (MCB-0234930) (RDM).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MLB and RDM conceived the study, supervised data collection and analysis and wrote the manuscript. ATP participated in data analysis and manuscript preparation. GHR participated in data analysis and data management. SI and AMN participated in data collection. KL-T provided access to the whole genome database needed for analysis. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2006_763_MOESM1_ESM.doc
Additional file 1: Summary of annotated Bandicoot ESTs. Table of bandicoot ESTs that match genes of known and unknown function in the public databases. Categories were assigned according to putative biological function using Gene Ontogeny and the Human Protein Reference Database [52, 53]. The closest protein match to each EST according to BLASTx is indicated. (DOC 928 KB)
12864_2006_763_MOESM2_ESM.doc
Additional file 2: Summary of integrated High Quality Non Coding (iHQNC) and inconsistent HQNC ESTs. Table of unmatched bandicoot ESTs that aligned to the opossum or human genomes or both. ESTs that aligned to the opossum or human genome, or both and were more than 5 kb from an annotated gene were labeled iHQNC ESTs. Those ESTs that aligned to both the human and opossum genomes, but were more than 5 kb from an annotated gene in only one species were labeled inconsistent HQNC ESTs. (DOC 189 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Baker, M.L., Indiviglio, S., Nyberg, A.M. et al. Analysis of a set of Australian northern brown bandicoot expressed sequence tags with comparison to the genome sequence of the South American grey short tailed opossum. BMC Genomics 8, 50 (2007). https://doi.org/10.1186/1471-2164-8-50
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-50