
Abstract
Both the hypoxia-inducible factor-1 (HIF-1) and tumor suppressor p53 are involved in the cellular response to hypoxia. How the two transcription factors interact to determine cell fates is less well understood. Here, we developed a network model to characterize crosstalk between the HIF-1 and p53 pathways, taking into account that HIF-1α and p53 are targeted for proteasomal degradation by Mdm2 and compete for binding to limiting co-activator p300. We reported the network dynamics under various hypoxic conditions and revealed how the stabilization and transcriptional activities of p53 and HIF-1α are modulated to determine the cell fate. We showed that both the transrepression and transactivation activities of p53 promote apoptosis induction. This work provides new insight into the mechanism for the cellular response to hypoxia.
Similar content being viewed by others


Introduction
Hypoxia, a decrease in oxygen availability, affects both physiological development and tumorigenesis1. A key mediator of the cellular response to hypoxia is the hypoxia-inducible factor-1 (HIF-1), which is a heterodimer of α and β subunits. HIF-1β is constitutively expressed, while HIF-1α is regulated in an oxygen-dependent manner2. HIF-1α is inactive and remains at low levels in normoxia. With enough oxygen available, the hydroxylation of HIF-1α by PHD (prolyl hydroxylase domain protein) promotes its degradation by pVHL (von Hippel-Lindau protein)3 and the hydroxylation of HIF-1α by FIH-1 (factor inhibiting HIF-1) represses its transcriptional activity via preventing the recruitment of co-activator p3004. In hypoxia, both PHD and FIH-1 are deactivated and thus HIF-1α is stabilized and activated. As a transcription factor, HIF-1 induces expression of target genes such as VEGF (vascular endothelial growth factor), EPO (erythropoiesis) and p215,6. VEGF and EPO regulate adaptive responses to hypoxia, while p21 induces cell-cycle arrest.
The tumor suppressor p53 also mediates the hypoxic response. p53 is kept at basal levels in unstressed cells because of Mdm2-mediated ubiquitination and proteasomal degradation. The p53 response to hypoxia is diverse, depending on the cell type, the degree and duration of hypoxia7. For example, p53 is stabilized via phosphorylation by the ATR (ataxia-telangiectasia mutated and Rad3-related) kinase only under severe hypoxia8. Moreover, high levels of p53 can induce apoptosis in a different manner from that when it is activated by DNA damage9,10. Notably, different mechanisms have been proposed for p53-mediated apoptosis in hypoxia: p53 promotes apoptosis mainly by transrepressing antiapoptotic genes like microRNA(miR)-17-9211,12, or by transactivating proapoptotic genes such as puma (p53-upregulated mediator of apoptosis), fas and bnip3l13,14,15. An issue naturally arises concerning whether these mechanisms are mutually exclusive or can be coordinated in one setting.
There exists an intricate interplay between p53 and HIF-1α. It was proposed that p53 stabilization is dependent on HIF-1α16; HIF-1α may bind to Mdm2, inhibiting Mdm2-dependent degradation of p5317. But HIF-1α accumulates with similar kinetics under mild and severe hypoxia, whereas p53 stabilization occurs only in severe hypoxia8. That is, HIF-1α upregulation is insufficient for p53 stabilization. On the other hand, it was reported that moderate p53 expression results in attenuated transcriptional activity of HIF-1 since p53 and HIF-1α compete for limiting p300, whereas high p53 expression leads to degradation of HIF-1α18. It is necessary to further clarify how the stabilization and transcriptional activities of p53 and HIF-1α are regulated under various hypoxic conditions.
Several mathematic models were developed to characterize the dynamics of the HIF-1 pathway, focusing on the mechanism for HIF-1α activation19,20,21. Few modeling studies explored the role for p53 in hypoxia. It is a challenge to reveal how the interplay between HIF-1α and p53 determines the cellular outcome. Here, we built a network model to characterize crosstalk between HIF-1 and p53 signaling, comprising their upstream modulators and downstream effectors. The analysis of network dynamics reveals that the cell either adapts to hypoxia or commits apoptosis, depending on the intensity of hypoxia. In mild hypoxia, HIF-1α accumulates to evoke transient cell-cycle arrest by inducing p21, whereas p53 remains at low levels. Under severe hypoxia, the accumulation of p53 attenuates HIF-1α activity and suffices to repress the expression of miR-17-92; consequently, BIM is induced to activate Caspase-3 and apoptosis ensues. In anoxia, p53 rises to high levels, transrepressing miR-17-92 and transactivating PUMA synergistically, whereas HIF-1α is degraded and inactivated. Thus, apoptosis can be triggered promptly. This work sheds new light on the mechanisms for the p53-HIF-1α interplay and hypoxia-induced apoptosis.
Modeling crosstalk between the HIF-1 and p53 pathways. We constructed an integrated model to explore the cell-fate decision upon hypoxia, characterizing the activation of HIF-1α and p53, their selective expression of target genes and apoptosis induction (Fig. 1). We focused on the interplay between HIF-1α and p53 under various hypoxic conditions. The key points of the model are presented as follows.

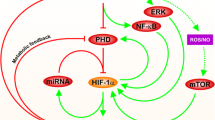
Schematic depiction of the model.
The model characterizes crosstalk between the HIF-1 and p53 pathways upon hypoxia. In hypoxia, HIF-1α is stabilized due to reduced hydroxylation by PHD. Under severe hypoxia, the ATR kinase is activated via auto-phosphorylation upon hypoxia-induced replication arrest and p53 is further activated by ATR. The shared coactivator p300 is required for the full transcriptional activity of p53 and HIF-1α. HIF-1α evokes transient cell-cycle arrest via inducing p21, whereas p53 can induce apoptosis via transrepressing or/and transactivating target genes. Dashed lines denote the expression of target genes by HIF-1α or p53, while solid arrowed lines represent the transitions between proteins. Circle- and bar-headed lines denote the promotion and inhibition of transition or production, respectively.
Regulation of HIF-1α activity. In normoxia, HIF-1α is kept at low levels due to oxygen-dependent hydroxylation that promotes its degradation by pVHL3. HIF-1α can also be degraded by Mdm2 in a p53-dependent manner22. Under hypoxic conditions, the hydroxylation of HIF-1α drops remarkably and HIF-1α accumulates. HIF-1α dimerises with HIF-1β in the nucleus. p300/CBP promotes the acetylation of HIF-1α, enhancing its transcriptional activity. In our model, HIF-1α is divided into unacetylated (HIF-1α, inactive) and acetylated (HIF-1αac, active) forms and their conversion is controlled by p300. The amount of p300 is limited18 and is set to a constant in the standard parameter setting. Because they can bind to p300 at different sites23,24, HIF-1α and p53 compete for limiting p300, the modeling of which is described later. Given the inhibitory effect of acetylation on protein degradation, only unacetylated HIF-1α is degraded through the PHD- or Mdm2-dependent mechanism22. The degradation and (de)acetylation processes are taken as enzyme-catalyzed reactions and assumed to follow the Michaelis-Menten kinetics (see Eqs. 1–2 in Supporting Material).
Activated HIF-1 can induce production of VEGF, EPO, PHD and p21. We explicitly characterize the induction of p21 and PHD with the Hill function (Eqs. 3 and 4), while implicating the roles for VEGF and EPO in the cellular adaptation to hypoxia. The HIF-1-PHD negative feedback promotes the adaptation of cells to mild hypoxia25,26. PHD is divided into inactive (PHD) and active (PHDa) forms and their conversion depends on oxygen concentration (Eq. 6).
Activation of ATR. In severe hypoxia, the ribonucleotide reductase activity declines, leading to production of single-stranded DNA (ss-DNA) at stalled replication forks. ss-DNA is then coated with replication protein A (RPA) and the ATR-ATRIP (ATR interacting protein) complex is recruited to ss-DNA, which promotes ATR phosphorylation at T198927. ATR phosphorylation enhances its interaction with TopBP1, which further activates the kinase activity of ATR. Thus, the activation of ATR is positively regulated.
ATR is divided into ATR (inactive) and ATRp (active) and their dynamics are described by Eqs. 7–8. The total level of ATR is assumed to be constant since ATR is mainly regulated posttranslationally27. The phosphorylation and dephosphorylation of ATR are characterized by the Michaelis-Menten kinetics28. The parameters are set to ensure that most of ATR is inactive in normoxia or mild hypoxia and ATR is quickly activated under severe hypoxia29.
Regulation of p53 activity. p53 is degraded by Mdm2, which is induced by p53. ATRp inhibits Mdm2 activity by phosphorylating it at Ser40730 and disrupts the p53-Mdm2 interaction by phosphorylating p53 at Ser15, thereby leading to p53 accumulation29. Based on its posttranslational modifications, nuclear p53 is divided into three forms: p53 (unphosphorylated), p53p (phosphorylated) and p53pac (phosphorylated and acetylated). Four forms of Mdm2 are defined here: Mdm2c (cytoplasmic unphosphorylated), Mdm2cp (cytoplasmic phosphorylated), Mdm2n (nuclear unphosphorylated) and Mdm2np (nuclear phosphorylated). For simplicity, we assume that only Mdm2cp can enter the nucleus31. Mdm2np cannot act as an E3 ubiquitin ligase30.
The coactivator p300 is required for the full transcriptional activity of p53 and HIF-1. Their competition for binding to p300 is characterized by the Michaelis-Menten kinetics with competitive inhibition (see Eqs. 1–2 and 10–11). The acetylation rate of p53p (or HIF-1α) is an increasing function of its own concentration and is a decreasing function of the level of HIF-1α (or p53p). On the other hand, p53, p53p and HIF-1α are targeted for degradation by Mdm2. The involved competition is also characterized by the Michaelis-Menten kinetics (Eqs. 1, 9 and 10). Notably, Mdm2-mediated degradation of p53pac is neglected since its dual modifications block their interaction. p53-induced expression of target genes, including mdm2 and puma, is all characterized by the Hill function and the Hill coefficient is set to 4 given the p53 tetramer acts as a transcription factor.
Mdm2c can be phosphorylated by Akt, promoting its nuclear entry32. Akt is activated via phosphorylation by PIP3 (phosphatidylinositol 3,4,5-trisphosphate), which is dephosphorylated by PTEN (phosphatase and tensin homolog) into PIP2 (phosphatidylinositol-4, 5-bisphosphate). The phosphorylation and dephosphorylation of Mdm2, Akt and PIP2/3 are all characterized by the Michaelis-Menten kinetics (Eqs. 12–15, 17 and 19). The total amount of Akt and that of PIP2 and PIP3 are separately assumed to be constant, similar to ref. 33.
Induction of apoptosis by p53. Different mechanisms have been proposed for p53-induced apoptosis in response to hypoxia. On one hand, repression of miR-17-92 by p53 is important for apoptosis induction since this relives some proapoptotic genes such as pten and bim from the inhibition by miR-17-9212,41. Our results suggest that reactivation of p53 could contribute to inhibition of HIF-1α activity.
The mechanism for p53 stabilization is somehow controversial in the literature. Previously, it was reported that p53 was stabilized by HIF-1α16. Later, this mechanism was challenged by the report that p53 accumulated only in severe hypoxia8. Thus, HIF-1α alone cannot stabilize p53. Rather, it was found that ATR contributes to p53 stabilization in severe hypoxia when replication arrest occurs. Here, we further show that the competition between p53 and HIF-1α for limiting p300 not only determines their transcriptional activity, but also regulates their abundance because p300-mediated acetylation prevents Mdm2-mediated protein degradation. p53 stabilization is enhanced when more Mdm2 is associated with unacetylated HIF-1α.
Our work may reconcile the contradictory reports on the mechanism for hypoxia-induced apoptosis11,12,13. We propose that depending on the severity of hypoxia, p53 may suppress the expression of antiapoptotic genes like miR-17-92, or inhibit miR-17-92 expression and activate PUMA expression synergistically to induce apoptosis. Accordingly, it takes a relatively long time to evoke apoptosis under severe hypoxia, or apoptosis is induced promptly after exposure to anoxia. Such an action mode of p53 is of functional significance, allowing for cellular adaptation to the hypoxic environment or eliminating cells promptly under strongly anoxic conditions. It would be interesting to experimentally test that.
We showed that effective repression of miR-17-92 expression is required for apoptosis induction under severe hypoxia. miR-17-92 itself can inhibit the production of BIM and PTEN34. Conversely, p53 transrepresses miR-17-92 to elevate BIM and PTEN expression12. BIM can induce apoptosis42, while PTEN contributes to p53 stabilization by sequestering Mdm2 in the cytoplasm32. It is worthy to explore whether other microRNAs induced by p53, such as miR-34a, contribute to apoptosis induction in hypoxia.
Finally, it is worth noting that the cellular response to hypoxia is extremely complicated. Here, we took a simplified approach to characterize the crosstalk between HIF-1α and p53 signaling by focusing on their competition in stabilization and transcriptional activity. We probed the effect of the severity of hypoxia on cell fate, while omitting the influence of the duration of hypoxia. Some other factors were also ignored, such as complex relationships between HIF-1, p53 and pVHL7 and different phosphorylation modifications of p53. Definitely, adding more facets of signal transduction to the model should shed new light on the hypoxic response.
Methods
The details of the model are presented in the Supporting Material. The concentration of each component is represented by a dimensionless state variable. The temporal evolution of dynamic systems is governed by ordinary differential equations, which are presented in Supplemental Method. All the initial values of variables are their lower steady-state values under normoxia and are listed in Supplemental Table S1. The standard parameter values are listed in Supplemental Table S2. Some parameters are set based on experimental measurements or known facts, while others are estimated by comparing the simulation results with experimental data. The unit of time is minutes, while the units of parameters are determined such that the concentrations of proteins are dimensionless. For simplicity, we do not indicate the units of the parameters explicitly. The rate equations are numerically solved using Oscill8.
The robustness of protein concentrations to parameter variations is also analyzed and the results are presented in Supplemental Table S3. The sensitivity of steady-state levels of  and p53pac to parameter variations is calculated when the parameter values are varied by ±10% with respect to their standard values. These concentrations are only sensitive to the parameters related to HIF-1α regulation, Mdm2-mediated protein degradation, or p53 production. Overall, our simulation results are robust to changes in most parameters and we provide a model for effectively exploring the interplay between p53 and
and p53pac to parameter variations is calculated when the parameter values are varied by ±10% with respect to their standard values. These concentrations are only sensitive to the parameters related to HIF-1α regulation, Mdm2-mediated protein degradation, or p53 production. Overall, our simulation results are robust to changes in most parameters and we provide a model for effectively exploring the interplay between p53 and  in response to hypoxia.
in response to hypoxia.
Additional Information
How to cite this article: Zhou, C.-H. et al. Modeling the interplay between the HIF-1 and p53 pathways in hypoxia. Sci. Rep. 5, 13834; doi: 10.1038/srep13834 (2015).
References
Semenza, G. L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56 (2010).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 92, 5510–5514 (1995).
Ivan, M. et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 292, 464–468 (2001).
Lando, D. et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471 (2002).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 (2003).
Greer, S. N., Metcalf, J. L., Wang, Y. & Ohh, M. The updated biology of hypoxia-inducible factor. EMBO J. 31, 2448–2460 (2012).
Sermeus, A. & Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2, e164 (2011).
Hammond, E. M., Denko, N. C., Dorie, M. J., Abraham, R. T. & Giaccia, A. J. Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 22, 1834–1843 (2002).
Zhang, X. P., Liu, F. & Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 108, 8990–8995 (2011).
Fridman, J. S. & Lowe, S. W. Control of apoptosis by p53. Oncogene 22, 9030–9040 (2003).
Koumenis, C. et al. Regulation of p53 by hypoxia: Dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol. Cell. Biol. 21, 1297–1310 (2001).
Yan, H. L. et al. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 28, 2719–2732 (2009).
Yu, J., Wang, Z., Kinzler, K. W., Vogelstein, B. & Zhang, L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 100, 1931–1936 (2003).
Liu, T. et al. Hypoxia induces p53-dependent transactivation and Fas/CD95-dependent apoptosis. Cell Death Differ. 14, 411–421 (2006).
Fei, P. et al. Bnip3L is induced by p53 under hypoxia and its knockdown promotes tumor growth. Cancer Cell 6, 597–609 (2004).
An, W. G. et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature 392, 405–408 (1998).
Chen, D., Li, M., Luo, J. & Gu, W. Direct interactions between HIF-1α and Mdm2 modulate p53 function. J. Biol. Chem. 278, 13595–13598 (2003).
Schmid, T., Zhou, J., Köhl, R. & Brüne, B. p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem. J. 380, 289–295 (2004).
Qutub, A. A. & Popel, A. S. A computational model of intracellular oxygen sensing by hypoxia-inducible factor HIF1α. J. Cell Sci. 119, 3467–3480 (2006).
Schmierer, B., Novák, B. & Schofield, C. J. Hypoxia-dependent sequestration of an oxygen sensor by a widespread structural motif can shape the hypoxic response - a predictive kinetic model. BMC Syst. Biol. 4, 139 (2010).
Nguyen, L. K. et al. A dynamic model of the hypoxia-inducible factor 1α (HIF-1α) network. J. Cell Sci. 126, 1454–1463 (2013).
Ravi, R. et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 14, 34–44 (2000).
Arany, Z. et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 93, 12969–12973 (1996).
Avantaggiati, M. L. et al. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89, 1175–1184 (1997).
D’Angelo, G., Duplan, E., Boyer, N., Vigne, P. & Frelin, C. Hypoxia up-regulates prolyl hydroxylase activity: A feedback mechanism that limits HIF-1 responses during reoxygenation. J. Biol. Chem. 278, 38183–38187 (2003).
Stiehl, D. P. et al. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels: Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 281, 23482–23491 (2006).
Liu, S. et al. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 43, 192–202 (2011).
Kholodenko, B. N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7, 165–176 (2006).
Hammond, E. M., Dorie, M. J. & Giaccia, A. J. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J. Biol. Chem. 278, 12207–12213 (2003).
Shinozaki, T., Nota, A., Taya, A. & Okamoto, K. Functional role of Mdm2 phosphorylation by ATR in attenuation of p53 nuclear export. Oncogene 22, 8870–8880 (2003).
Mayo, L. D. & Donner, D. B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 98, 11598–11603 (2001).
Manning, B. D. & Cantley, L. C. AKT/PKB signaling: Navigating downstream. Cell 129, 1261–1274 (2007).
Wee, K. B. & Aguda, B. D. Akt versus p53 in a network of oncogenes and tumor suppressor genes regulating cell survival and death. Biophys. J. 91, 857–865 (2006).
**ao, C. et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 9, 405–414 (2008).
Kloet, D. E. A. & Burgering, B. M. T. The PKB/FOXO switch in aging and cancer. BBA-Mol. Cell Res. 1813, 1926–1937 (2011).
Cory, S. & Adams, J. M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–56 (2002).
Kirsch, D. G. et al. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 274, 21155–21161 (1999).
Levine, A. J. p53, the cellular gatekeeper for growth and division. Cell 88, 323–331 (1997).
Schofield, C. J. & Ratcliffe, P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354 (2004).
Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers and tumor-associated macrophages. Am. J. Pathol. 157, 411–421 (2000).
**a, Y., Choi, H. K. & Lee, K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 49, 24–40 (2012).
Bouillet, P. et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis and to preclude autoimmunity. Science 286, 1735–1738 (1999).
Acknowledgements
This work was supported by the 973 program of China (No. 2013CB834104), National Natural Science Foundation of China (Nos. 11175084, 11204126 and 31361163003) and PAPD.
Author information
Authors and Affiliations
Contributions
X.P.Z. and F.L. designed research; C.H.Z. performed numerical simulations; C.H.Z., X.P.Z., F.L. and W.W. analyzed data; C.H.Z., X.P.Z. and F.L. wrote the paper; F.L. coordinated the research. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhou, CH., Zhang, XP., Liu, F. et al. Modeling the interplay between the HIF-1 and p53 pathways in hypoxia. Sci Rep 5, 13834 (2015). https://doi.org/10.1038/srep13834
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13834
- Springer Nature Limited
This article is cited by
-
Preliminary verification of the anti-hypoxia mechanism of Gentiana straminea maxim based on UPLC-triple TOF MS/MS and network pharmacology
BMC Complementary Medicine and Therapies (2022)
-
How microcompetition with latent viruses can cause α synuclein aggregation, mitochondrial dysfunction, and eventually Parkinson’s disease
Journal of NeuroVirology (2021)
-
The regulatory roles of p53 in cardiovascular health and disease
Cellular and Molecular Life Sciences (2021)
-
How an increase in the copy number of HSV-1 during latency can cause Alzheimer’s disease: the viral and cellular dynamics according to the microcompetition model
Journal of NeuroVirology (2021)
-
NLRP3 Inflammasome is Activated in Rat Pancreatic Islets by Transplantation and Hypoxia
Scientific Reports (2020)