Bioinformatic pipeline to detect transfer events
The xenoseq pipeline (https://github.com/bramvandijk88/xenoseq, Fig. 1c–e) is a wrapper that combines read trimming, assembly, read map**, read filtering, and local alignment to seek evidence of horizontal gene transfer or the transfer of other nanoscale entities between evolving communities. This pipeline takes as input raw (untrimmed) fastq files containing shotgun metagenomic data of derived samples (query, in which to search for newly introduced sequences) and datasets from ancestral samples (reference). Query reads are trimmed using fastp [57] (v0.23.2), mapped against the corresponding reference contigs using BWA [58, 59] (v0.7.17) using default options, and samtools [60] (v1.15.1) is used with the flag ‘−4’ to extract unmapped reads. These reads are assembled into ‘unique contigs’ using Megahit [61] (v1.2.9) (xenoseq_find, Fig. 1c). Next, these unique contigs are blasted against a local database using NCBI Blast [62] (v.2.13.0) of all other reference communities to link the emergence of unique contigs to allopatric ‘donor’ communities. Linked contigs are extracted from unique contigs using Seqkit [63] (v.2.3.0) (xenoseq_link, Fig. 1d). By default, contigs are linked to a donor when blast has at least one high-scoring segment with a minimum length of 300 with at least 99% nucleotide identity. While the algorithm and cut-offs limit the sensitivity over larger evolutionary distances, they are designed to detect sequences that have recently diverged and are still largely identical. The contigs that match target sequences in allopatric communities are referred to as ‘xenotypic contigs’. Finally, to detect shifts in abundance of the detected contigs, reads from all query and reference samples are mapped (xenoseq_trace), and coverage/breadth statistics are saved in a tab-separated file. This allows for visualisation of transfer across communities (Fig. 1e). In this manuscript, we further analyse sequences >10 kb in length.
By default, xenoseq runs all subroutines (xenoseq_find, xenoseq_link, xenoseq_trace), but each subroutine can be run independently by modifying the command-line flags. Finally, xenoseq uses GNU parallel59 to run multiple jobs simultaneously.
Xenoseq benchmark
To benchmark the pipeline, introduction of MGEs into mock-communities was simulated (see Supplementary Material I full details). Six bacterial genome sequences were downloaded from RefSeq. Two mock-communities were then generated, one with an even taxon distribution (easy dataset), and one with a highly skewed taxon distribution (hard dataset). Simulated MGE sequences were either i) randomly inserted into genome sequences, representing integrative elements, or ii) included as separate replicons linked to a single host genome by addition to the genome fasta file as a separate contig. Illumina sequencing was simulated from the resulting ‘communities’ with ART62, using both default and ten-fold elevated error rates, which were used as input to benchmark the pipeline. Simulation of mock-communities and horizontal gene transfer events was done in R v4.1.3 using the packages biostrings60 v2.62.0 and seqinr61 v4.2.16. Another (small) mock-community dataset is available on the repository, and can be used to rapidly test whether the pipeline and its dependencies are configured correctly.
Metagenome-assembled genomes
We generated MAGs from all twenty compost communities. By combining samples across multiple time points, we improved the potential detection of rare types whose coverage in a single sample is insufficient for assembly. Quality control of sequencing reads was done with Prinseq version 0.20.4 [64] using ‘–derep 14 -lc method -c threshold 20’. We trimmed adapters with Flexbar v.3.5.0 [65] using the ‘–adapter-preset Nextera -ap ON’ flag. Combined reads from multiple time points for each community were cross-assembled de novo using metaSPAdes v.3.14.0 [66]. Reads from each sample were mapped back to the assembled contigs using bwa-mem v.0.7.17-r1188 [59], and coverage calculations were performed with SAMtools v1.7 [60]. Contigs were binned using metaBAT2 v2.12.1 [67] and MAG quality was assessed with CheckM v1.1.3 [68]. All MAGs and contigs were annotated using BAT and CAT (Bin/Contig Annotation Tool, respectively, v.5.1.2) [69], which uses prodigal [70] to predict open reading frames and diamond [73] by extracting 16S sequences with Barnapp 0.9, and a neigbour-joining tree was created with Geneious 2023.1.2.
DNA degradation assay
To investigate whether the identified sequences are derived from living cells or DNA liberated by lysed cells, we tested the ability of microbial compost communities to degrade extracellular DNA. To do this, four experimental microbial communities were established from samples of four independent compost heaps. Compost was washed in M9 salt solution and stored with glycerol saline at −80 °C. Frozen stocks were thawed on ice and 4.25 mL of the stock was then washed of glycerol by two successive cycles of centrifugation (4000 g, 10 min) and resuspension (in 5 mL sterile M9 salt solution), followed by a final resuspension in 1 mL M9 salt solution. The washed cells were then added to 100 mL bottles with 19 mL M9 media and a piece of cellulose paper as a carbon source. Mesocosms were incubated without shaking at 28 °C for 14 days to allow community growth. The entire volume including any remaining paper was then transferred to 50 mL centrifuge tubes, vortexed for five minutes to produce a slurry, and 1 mL of slurry was transferred to fresh M9 media for another 14 days of incubation. During this time a stock of genomic DNA (gDNA) from Escherichia coli (REL606) was made using the ‘DNeasy Ultraclean Microbial kit’ (Qiagen), resulting in a stock of ~30 mL of gDNA at 20 µg/mL (stored at −20 °C).
Communities were then propagated a second time to four replicate cellulose-mesocosms spiked with gDNA. Community-free mesocosms were also established which acted as controls. All mesocosms were spiked with ten µg of E. coli gDNA, and were destructively harvested after 0, 1, 2 and 14 days of incubation. For sampling, the contents of each mesocosm was transferred to a 20 mL centrifuge tube, reduced to a slurry and then centrifuged at 4000 g for 5 min. The supernatant was transferred to 15 mL sterile tubes and frozen at −80 °C for later DNA extraction. In total, four replicate communities and three community-free controls were harvested at each time-point.
To measure relative DNA degradation by communities over the time-course, supernatant samples were thawed on ice, and further spiked with an internal standard of one µg/mL of gDNA of Pseudomonas fluorescens SBW25. Samples were then vortexed and samples were filtered using a 0.2 µm syringe-filter. DNA of 2 mL of spiked filtrate was isolated using a Phage DNA Isolation Kit (Norgen Biotek Corp) following the manufacturer’s instructions, except for loading two (rather than one) mL sample through a single purification column to improve DNA yield. Extracted DNA was then sequenced using a NextSeq MidOutput 300 cycle run resulting in 2 × 150 bp paired-end reads (see Supplementary Table I). Reads were aligned to the non-redundant (NR) protein database [72] with Diamond54 using the blastx subroutine, where the top hit was used to identify the read as being either P. fluorescens, E. coli, or ‘other’ (the latter representing either species present in the communities or erroneously assigned reads).
MGE classification
Viral sequences were predicted using Vibrant (v1.2.0) [44], Virsorter2 (v2.2.3) [45], Seeker (v1.0.3) [46], and CheckV (v0.8.1) [74]. While CheckV is primarily designed to estimate completeness of candidate viral genomes, we found that sequences with either low, medium or high scores were typically identified as viral by other tools as well (see Fig. 3 in main text). Plasmid sequences were predicted using PlasFlow (v1.1.0)w [47] and PlasClass (v0.1) [48] and, if relevant, the origin of replication with OriFinder 2022 [75]. ICEs were identified with ICEfinder [49]. IS elements were predicted with ISEscan (v1.7.2.3) [1e).
Note that xenoseq can in principle be applied to datasets that deviate from this particular experimental design. While Quistad et al. use glass mesocosms, mice would equally suffice when one is interested in the evolution of gut microbiomes. The experimental design could reveal selection pressures experienced by such natural communities, where the movement of MGEs (and the genes they carry as cargo) reflects what traits are relevant for community function under those conditions. Additionally, one could expose evolving communities to particular selection pressures, e.g. for antimicrobial resistance, to discover new MGEs that encode traits relevant under that condition. The only requirement is that longitudinal samples are taken from parallel communities which undergo exchange of MGEs or other biologically relevant entities.
Horizontal compost mesocosms are enriched in xenotypic sequences
The mesocosms from the study by Quistad et al. represent a ‘challenging case’ for the xenoseq pipeline, because of the unprecedented diversity and abundance of rare types. As mentioned above, shifts in abundance of rare types (as observed by Quistad et al.) can generate false positives (Fig. 1b). As a control, we therefore ran xenoseq on vertical communities, which are ‘closed’ and unaffected by movement of MGEs from allopatric communities. Indeed, we found that unique sequences are present in both horizontal and vertical communities (Fig. 2a, a total of 5617 and 6883 sequences respectively). However, only horizontal communities contained unique contigs that could be linked to allopatric communities (Fig. 2b). In total, 1756 (31.2%) of contigs in horizontal communities could be linked to an allopatric ‘donor’ community, versus only 58 (0.8%) of contigs from vertical communities. The remaining 58 contigs may be spuriously linked to allopatric communities due to overlap between ancestral compost communities. While all derived compost communities showed evidence of DNA sequences that were amplified over time, evidence for allopatric origins of these sequences was found exclusively in horizontal communities.
A possible confounding factor in the detection of xenotypic sequences arises if naked DNA included in the MGE cocktail were to remain intact during the 14-day incubation period thus ending up in the metagenomic data. To test this, we added exogeneous gDNA from Escherichia coli to the communities and monitored its persistence over time (see Methods). In the presence of the microbial communities, E. coli gDNA was no longer detectable within 48 h after addition (Supplementary Fig. 3), while it remained detectable throughout the 14-day experiment in community-free controls. Furthermore, with DNA concentrations of 0.67 ng/ml in the initial MGE cocktails, we consider it highly unlikely that these sequences would significantly contribute to the community metagenome samples after 14 days. Taken together, these data demonstrate that in order for xenoseq to predict horizontal transmission, DNA sequences must first persist degradation, and then be sufficiently amplified within the recipient community. Such amplification indicates some form of selection for the candidate MGE, e.g. because it is a selfish element or conveys a substantial fitness benefit to a host.
Xenotypic sequences are enriched for phages and IS-elements
The MGE cocktail that was distributed among mesocosms could contain bacteriophages, plasmids, naked DNA, membrane vesicles, and potential other (unknown) vehicles of transfer. The 1756 xenotypic sequences were interrogated using a variety of MGE detection tools, which provided predictions as to whether the sequences are viral [44,45,46, 74], plasmid45,46, IS-carrying49, ICE-carrying47, or integron-carrying48. We found that 714 (40.6%) of xenotypic sequences were identified by least one MGE prediction tool (see Fig. 3a). Considerable overlap was found between phages and plasmids (Fig. 3b), but also between other elements. These results indicate the existence of recombined or hybrid MGEs like phage-plasmids29. They may also highlight challenges of unambiguously predicting the MGE type from sequence using the selected tools.
To test whether xenotypic sequences are enriched in MGEs, these data were compared to an arbitrary set of sequences with a similar length distribution, sampled from the metagenomic contigs. Note that these community samples are still expected to contain many MGEs, but perhaps less than the xenotypic sequences. Indeed, phages and IS-elements appear to be over-represented among xenotypic sequences, whereas the numbers of plasmids, ICEs, and integrons are not significantly different (Fig. 3c). When corrected for multiple testing, phages remained significantly overrepresented but this did not hold for IS elements (see Supplementary Table I, Sheet 3 for all p values). In summary, because canonically selfish elements appear enriched, the results may further corroborate the importance of sequence amplification after introduction into a new community.
Interestingly, after applying nine state-of-the-art tools for predicting five categories of MGEs, 1042 xenotypic contigs (59.4%) remained unidentified.
Movement of xenotypic sequences between communities
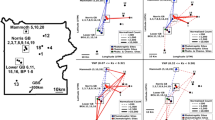
To investigate the dissemination and dynamics of xenotypic contigs, xenoseq was used to map reads from all communities to the xenotypic contigs representing candidate MGEs. Figure 4 shows the results for seven selected examples: a complete 200 kb phage (Fig. 4a), an incomplete but abundant phage sequence (Fig. 4b), a putative viral sequence exclusively predicted by Seeker (Fig. 4c), an element predicted as both phage and plasmid unanimously (Fig. 4d), a large 313 kb plasmid (Fig. 4e), and a stretch of apparent chromosomal DNA adjacent to an IS3 element (Fig. 4f).
Xenoseq also revealed transfer of chromosomal bacterial DNA fragments lacking MGE predictions, many of which were annotated as Candidatus Saccharibacterium (Fig. 4g), a member of the candidate phylum radiation (CPR) bacteria first observed in human oral cavities [85]. Although this nanobacterium could indeed be sufficiently small to pass through the 0.2 µm filter, members of this species appear in both horizontal communities (community 1, 3, 4, 5, 7, 8, 9, and 10) as well as vertical communities (community 1, 3, 4, 7, 8, 9, 10), with varying estimates of genome completion (see Supplementary Table 1). The best representative MAG of this species was observed in HC10, which has a small genome (818 kb) that was nevertheless estimated to be 97% complete, and indeed encoded many of the relevant housekee** genes (e.g. gyrA, recA, polA, topA, rpoB, and a single 16S ribosomal RNA gene). Comparing the 16S sequences with those from recently published CPR bacteria and DPANN archaea from groundwater samples [73], shows it clusters together with other CPR bacteria annotated as Candidatus Saccharibacterium (see Supplementary Fig. 4). Interestingly, alignment-free estimates of average nucleotide identity (fastANI v1.33) revealed that the (near-complete) nanobacterial MAGs from the communities are highly similar (up to 99.4% ANI). However, fastANI with respect the groundwater CPR bacteria yielded no results, which occurs when the genomes compared are too dissimilar. Taken together, these analyses suggests that the nanobacterium in our compost mesocosms are closely related to, but distinct from, these previously published bacterial MAGs.
Co-occurrence analysis (see Supplementary Fig. 5a) further reveals a potential connection between the identified nanobacterium and the genus Cellvibrio. Tracking the abundance of both players over times suggests a potential boom-and-bust dynamic, punctuated by and long periods of stasis (Supplementary Fig. 5b). Such dynamics may be indicative of a pathogenic lifestyle.
Compost mesocosms form distinct community types
Thus far, we have illustrated that treating evolving microbial communities with an ‘MGE cocktail’ derived from all communities promotes the movement of various MGEs, and even nanobacteria between communities. We have also shown that xenotypic contigs are especially enriched in SGEs, suggesting the importance of independent sequence amplification for the survival of MGEs after introduction into allopatric communities. In the following sections, we examine the ecological and evolutionary consequences of this treatment. To investigate whether and how the horizontal communities are distinct from the vertical communities, we studied the abundance of MAGs. These MAGs were assigned a taxonomic classification using the Bin Annotation Tool (BAT). When taxonomic rank could not be determined reliably due to conflicting ORFs, a higher-order taxonomic rank was assigned instead, ensuring all MAGs had a robust classification.
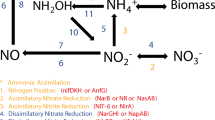
Each MAG was screened for various genes related to two metabolically relevant functions: cellulose degradation and nitrogen metabolism (see Methods and Supplementary Table I, Sheet 5). The relative abundance of dominant MAGs is shown for each community in Fig. 5a (for an interactive graph of all MAGs, see Supplementary Files). The MAGs are dominated by either Rhodanobacter (shown in brown in Fig. 5a) or Cellvibrio lineages (shown in green). Members of both these genera are able to degrade cellulose, the sole carbon source in the mesocosms (Fig. 5b). As many other MAGs do not have this ability, Rhodanobacter and Cellvibrio appear to occupy a similar niche and are the primary degraders of cellulose, which could explain why they appear to be mutually exclusive. The two community types furthermore have distinct lineages that coinhabit the mesocosms. For example, the communities dominated by Rhodanobacter lineages often coexist with Nitrosomonas europaeae (steel blue), which can reduce nitrate and nitrite (Fig. 5b). Communities dominated by Cellvibrio instead frequently host species of above-mentioned C. Saccharibacteria (denoted by an asterisk in Fig. 5a, b.
Horizontal communities show more changes in primary degraders
As can be seen from Fig. 5a, vertical communities (VCs, left) are relatively stable in the composition of dominant MAGs, establishing either with Cellvibrio or Rhodanobacter as primary cellulose degraders. Horizontal communities (HCs) however show rapid shifts in these primary degraders. For example, HC2 initially establishes with Rhodanobacter (brown) as the primary degrader. However, during week 4 to 8, a Cellvibrio lineage (C1, green) becomes more dominant relative to other MAGs. Finally, Rhodanobacter re-emerges as the primary degrader. Vice versa, HC8 and HC9 are initially established as Cellvibrio-communities, and show a transient appearance of a Rhodanobacter lineage after 32/40 weeks.
While the above-mentioned disruptions caused by the MGE cocktails are transient, Cellvibrio C1 in HC6 appears to completely overturn the ecosystem structure that was established during the preceding 32 weeks, although it is unclear whether this disruption is transient since we do not have measurements after 48 weeks. Cellvibrio C1 also emerges in three other mesocosms independently (in HC9 at week 8, and HC4 and HC10 at week 32), and its emergence is always accompanied by a MAG containing only a single 313 kb contig (shown in purple in Fig. 5a, henceforth called Cp). This contig is of circular nature, and is in fact the same plasmid-like element earlier identified by xenoseq (Fig. 4e). The large mobile element carries plasmid partitioning proteins (e.g. ParB and ParM) typically associated with low copy-number plasmids [86, 87] (see Fig. 5c), which is consistent with it co-occurring with Cellvibrio C1 in an approximate 1:1 ratio. In addition to features that are akin to large plasmids, Cp also carries many ORFs associated with phages as evident in matches to the viral PHROGs-database66 (for example, integrase and endolysin). Finally, Cp carries a putative conjugative region identified by ICEfinder. As this plasmid is present in earlier time points only in VC/HC8, this suggests this community as the donor. We hypothesise that Cp transfers horizontally, enabling Cellvibrio C1 (the putative host) to displace the previously established ecosystem structure.
Ammonia production appears elevated in Cp+ communities
Because cellulose is the only exogenously provided carbon source in the mesocosms, the ability to degrade cellulose is not surprisingly found in many MAGs (see Fig. 5b). While all the Cellvibrio lineages have the ability to degrade cellulose, one particular Cellvibrio (C1) is unique in also carrying a nitrogen fixing enzyme (flavoprotein, EC 1.19.6.1). The ability to fix nitrogen is likely important, because 1 mM ammonium chloride from the M9 medium added every 14 days is the only source of exogenously supplied nitrogen. We hypothesised that the Cp plasmid may have regulatory impacts on C1 host metabolism, perhaps favouring fixation of nitrogen.
In the study by Quistad et al., ammonia production was measured at the end of the experiment (T = 48 weeks). By plotting the abundance of Cellvibrio C1 against data on ammonia production, a strong positive correlation was observed (Fig. 6a) in horizontal communities, but much less so in vertical communities. Note that C1 in most horizontal communities carries the Cp plasmid, and C1 in vertical communities does not. We suggest that this indicates that C1 may be responsible for the accumulation of ammonia, but only in the presence of the Cp plasmid. Indeed, by taking the slope of the linear regression, we found that horizontal communities show a significantly steeper correlation between C1 and ammonia production rates, especially early in the growth cycle (Fig. 6b). Finally, when focussing only on horizontal communities where Cp was absent (HC3 and HC7), no positive correlation was observed (Supplementary Fig. 6). These data suggest that Cp promotes the fixation of nitrogen by Cellvibrio C1.